两阶段设计在医疗器械非随机临床试验中的应用
2018-01-03赵延延许毓君
赵延延 许毓君 王 杨 李 卫△
两阶段设计在医疗器械非随机临床试验中的应用
赵延延1†许毓君2†王 杨1李 卫1△
目的介绍两阶段设计的概念、使用流程和注意事项,并以某外周血管支架临床试验为例介绍其在医疗器械非随机临床试验中的应用。方法基于倾向性评分的两阶段设计能使医疗器械非随机临床试验满足随机化和前瞻性的原则。我们以某外周血管支架的非劣效试验为例,介绍两阶段设计的流程和细节。结果两阶段设计解决了非随机临床试验由于非随机所带来的可能的研究偏倚及倾向性评分方法使用过程中的主观不确定性,实现了对随机临床试验的模拟,从而保障了非随机临床试验研究设计和统计分析的客观性和前瞻性。结论两阶段设计能够增加临床试验的可行性,整合高质量的外部数据,缩短试验周期,提高试验效率,最终得到可靠的结论,具有较大的推广价值。
两阶段设计 倾向性评分 医疗器械 非随机临床试验
设计科学、实施严谨的随机对照试验是评价医疗产品安全性和有效性的金标准。然而出于伦理学或临床可行性的考虑,许多医疗器械临床试验无法采用严格的随机对照研究设计[1];此外,随机对照试验往往耗时很长,如医疗器械临床试验的周期是3~7年,而随着医疗技术的发展,器械产品的更替周期要短于随机对照试验的周期,如心脏支架产品的生命周期约为2年。为了能够使好产品尽早上市,造福于广大病患,急需一种既能够作为随机临床试验补充,又符合统计学规范的研究设计方法。
非随机临床试验(non-randomized clinical trial)是指在临床试验中受试者所分配的干预不是由机会决定,而由受试者或研究者指定[2],根据对照组的不同可以分为非随机同期对照试验和非随机历史对照试验。相比于传统的随机对照试验,这类试验能够克服实际研究开展过程中面临的伦理学或可行性问题,整合高质量的外部数据,高效地回答临床实践问题,在国外已有不少被用于医疗器械上市前的申报中[3-4]。但非随机临床试验本身缺少随机化的环节,会导致组间基线变量不均衡,带来效应估计的偏倚;同时结局评价不具有前瞻性,即研究者在获得试验结局数据之后再进行评分建模,通过大量重复的事后分析得到有利于试验产品的结果,这使得试验结果的可靠程度备受质疑,目前在国内尚无以非随机对照临床试验的结果作为上市前确证性证据而获得审批的产品。倾向性评分的方法本身虽然在解决随机化问题时显示出极大的优越性[5],但在临床试验中多被用于事后分析,存在数据导向、人为操控数据获得阳性结果的嫌疑,分析结果通常亦不被认可。
两阶段设计是指在利用倾向性评分方法的基础上,对试验的流程进行人为的划分[6],从而模拟试验的随机化过程,保障试验设计和统计分析的前瞻性,进而获得较为客观可靠的试验结果,被美国食品药品监督管理局(FDA)推荐用于非随机临床试验中[7]。本文将详细介绍两阶段设计的基本原理和在医疗器械临床试验中的应用,并以某外周血管支架产品安全性和有效性评价为例,阐述其使用流程和注意事项。
基本原理
两阶段设计的本质是实现非随机临床试验对随机对照临床试验随机化和前瞻性的模拟。随机化保证了观测到和未观测到的变量在试验组和对照组间的分布是均衡的,进而依据因果推断的原理得到效应的无偏估计。前瞻性原则,即在方案设计阶段研究者无法提前接触到结局数据,使得试验结果客观可靠[8-9]。
1.倾向性评分与随机化原则
非随机临床试验的基线变量若未经处理,往往存在组间不均衡的情况。这些不均衡变量的维数很高且不少为连续型变量,此时观察性研究中常用的混杂校正方法如匹配、分层、回归并不适用:匹配能够校正的因素非常有限;即便进行了分层,当每层的人数很少甚至没有时,无法进行效应估计;多因素校正则要求有足够数量的结局事件,而许多临床试验并不满足条件。此时随机化的效果可以通过倾向性评分来实现,该方法最早由Rosenbaum和Rubin于1983年提出[5],在2002年被美国FDA采纳用于医疗器械上市前的非随机临床试验及药物和器械上市后的监管研究中[10]。
倾向性评分定义为在观察到的协变量(Xi)条件下,研究对象i(i=1,2,…,n)被分配到试验组(Zi=1)而非对照组(Zi=0)的条件概率,可表达为:
e(Xi)=P(Zi=1|Xi)
该方法基于降维的思想,当模型中纳入了全部的混杂变量时,每个试验对象的基线信息便能够被整合至一维的倾向性评分中,可通过logistic回归模型实现。进一步地,只需对倾向性评分进行分析,如匹配、分层、回归或逆概率加权等,便能同时均衡两组间全部混杂变量,实现随机化的模拟。
2.两阶段设计与前瞻性原则
在非随机临床试验中,为确保倾向性评分估计与最终的结局分析独立进行,避免前瞻性原则遭到破坏,需要利用两阶段设计(two-stage design)[7-8]对试验流程进行人为的划分,如图1所示。
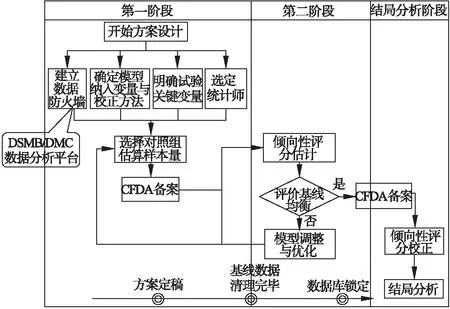
图1 两阶段设计的基本流程
两阶段设计包含两个层次,首先试验被严格地分成方案设计阶段(design phase)和结局分析阶段(analysis phase)。方案设计阶段主要包括倾向性评分建模、模型评价和调整、制定和完善统计分析计划等步骤,在此期间试验正式开展,但所有结局数据(特别是当采用外部数据作为对照时)均需由防火墙隔离,在整个方案设计阶段统计师都无法获得。当数据库锁定,进入结局分析阶段,倾向性评分模型不得更改,统计师在校正倾向性评分的基础上对结局数据进行分析,得到最终统计分析结果。数据防火墙的建立是划分方案设计阶段与结局分析阶段的关键,也是两阶段设计在实际应用中的最大挑战,可通过第三方机构如数据安全监查委员会(DSMB/DMC)[11]的介入、数据分析平台的访问权限控制、留痕功能[12]或其他方式实现,但无论采用何种方法,都必须在制定方案之初与临床试验法规监管部门充分沟通达成一致。
在此基础上,方案设计阶段被进一步分为第一阶段和第二阶段。从开始方案设计到试验正式开始为第一阶段,主要任务包括选定独立的统计师,确定采集的基线变量,选择适合的对照组和估算样本量。另外,需要在方案中明确倾向性评分模型纳入变量的原则、组间平衡的评价标准和结局分析阶段校正评分的方法,在方案定稿并交由CFDA备案后,这些信息便不可更改。当完成受试者入组与基线信息采集,基线数据清理完毕,第一阶段即完成,进入第二阶段。统计师进行倾向性评分估计,根据评分进行匹配或者分层,对组间基线变量的均衡情况进行评价。当两组评分的重叠部分过小时,可能需要对模型进行调整和优化,直至两组基线变量均衡可比,并最终完善统计分析计划。研究者在方案设计阶段应当与法规监管部门进行充分沟通,并于第一阶段和第二阶段结束后及时进行备案,缺少这些环节将严重影响最终试验结果的可靠性。
实例分析
选取真实临床试验中的部分数据作为案例,对两阶段设计的具体流程加以阐述。某外周血管支架新一代产品的非劣效临床试验,假设试验组与对照组主要终点指标术后12个月无靶血管或靶病变再次血运重建发生率均为92%,非劣效界值δ为-5%,统计检验的显著性水平(α)取单侧0.025,考虑脱落率10%,当每组受试者525例时,得到非劣效结论的检验效能(1-β)超过80%。本案例中由于试验支架的前代产品已上市,两代产品相似度高,适用人群一致,且前代产品的随机对照试验开展时间接近,积累了高质量的试验数据,故选择这部分数据作为本次试验的对照。研究者对于试验数据的全部操作均在可留痕的数据分析平台上进行,并根据不同的角色分工和试验的不同阶段给予不同的平台访问权限。同时,在方案中明确了试验需要采集的关键变量,入选及排除标准等信息。基于临床经验及既往文献,在第一阶段我们确定了12个对主要终点指标有影响的混杂变量。此外,倾向性评分模型还将纳入组间有显著差异的基线变量。倾向性评分采用1:1最邻近法(nearest neighbor)进行匹配,当模型调整后标准化均数差小于0.1时认为变量在组间分布均衡,在结局分析阶段倾向性评分通过分层进行校正。
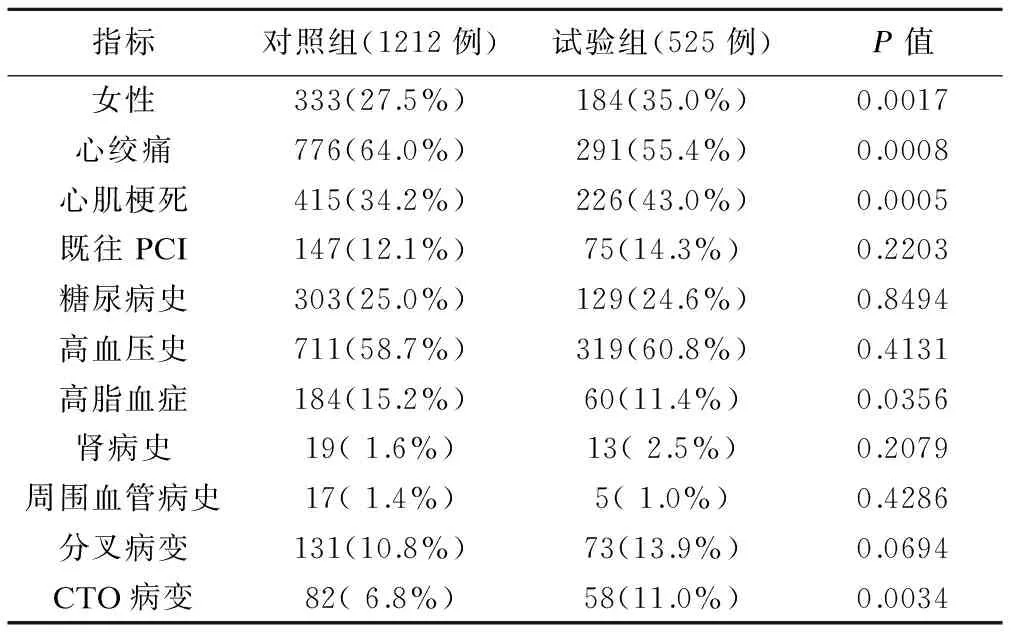
表1 试验组和对照组纳入倾向性评分模型基线定性变量比较

表2 试验组和对照组纳入倾向性评分模型基线定量变量比较
表1及表2展示了最终纳入模型的21个基线变量在试验组和对照组的分布情况。选择logistic回归模型估计两组的倾向性评分,并对评分进行匹配,匹配后试验组和对照组各525例。比较分析匹配后的基线变量,得到标准化均数差均小于0.1(图2),说明匹配后的基线指标组间均衡。
在结局分析阶段,对匹配后的人群进行倾向性评分分层(表3),分别在每层进行比较并通过CMH卡方计算总效应估计值,试验组术后12个月无靶血管或靶病变再次血运重建发生率为94.48%,对照组为96.76%,率差(试验组-对照组)为-1.76%(95%CI:-4.09%~0.63%),由于下限-4.09%大于方案中预先设定的非劣效界值-5.0%,可以认为试验组的有效性非劣于对照组(P=0.007)。
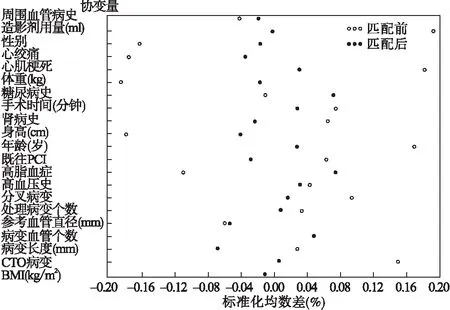
图2 倾向性评分调整前后基线变量的标准化均数差
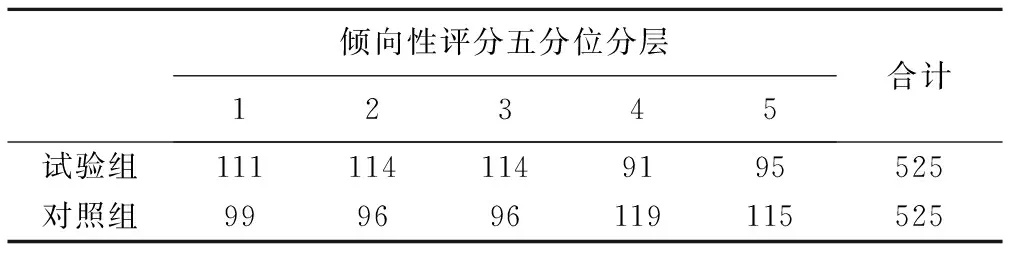
倾向性评分五分位分层12345合计试验组1111141149195525对照组999696119115525
讨 论
由于医疗器械随机对照试验的特殊性,在实际开展过程中往往会面临不少伦理学和可行性问题。对于市场上已有成熟同类产品的试验产品,当知情同意获取困难、受试者可能不同意随机分配等情况出现时,非随机临床试验可以作为随机对照试验的补充,从一定层面上提供产品的安全性和有效性证据。此外,非随机临床试验具有控制时间和经济成本、充分利用高质量外部数据等优势。两阶段设计为非随机临床试验提供了严谨的流程规范。本文在介绍两阶段设计原理的基础上,以实际临床试验数据为案例,阐述了其在医疗器械非随机临床试验中的具体使用步骤。
在实际运用两阶段设计时,应当注意如下问题:
1.从监管角度而言,建立合理的防火墙机制,避免已有的结局数据发生泄漏、统计师在获取结局数据后进行重复分析是确保非随机临床试验前瞻性的关键。通过第三方机构如DSMB/DMC介入、数据分析平台的访问权限控制与留痕功能来实现前期的数据保密是可取的,此外也可以采用其他方式建立数据防火墙,但均需要在试验方案中写明,并于试验正式开展前与临床试验法规部门进行沟通、达成一致。
2.两阶段设计对非随机临床试验的流程进行了严格的划分,当某阶段结束后,方案中早已明确的倾向性评分模型纳入变量原则、结局评价过程中评分校正方法等信息便不能更改。因此在整个试验期间,研究者需要与临床试验法规部门进行多次沟通,并在每一阶段结束后及时备案,否则将严重损害试验的可靠性,导致最终结果不被认可。
3.对照组数据可以是已上市同类产品的同期试验数据、同类产品的既往临床试验数据或注册登记研究,其中同期试验数据可靠性最高。若采用既往临床试验数据,应当注意两组人群的入组时间应尽可能接近,入选和排除标准尽量一致,否则指南的更新、医疗实践和技术的发展都会造成人群选择和临床结局定义及判断的差异,影响数据可比性。此外,关键变量缺失会造成潜在的混杂效应无法处理,增加结果评价的不确定性,因此在方案设计第一阶段应当明确需要采纳的变量,选择包含关键变量的对照组数据。
4.当两组数据可比性不高时,在校正倾向性评分的过程中试验样本量会发生较大变化,检验功效亦会受损。因此在方案设计的第二阶段,统计师应当基于当前的倾向性评分模型对样本量和检验功效进行重新估计,并在统计分析报告中写明。
5.对于随访周期很短或被试产品的主要评价指标为即刻终点的临床试验,由于较难进行明确的流程划分,因此不建议采用倾向性评分的两阶段设计方法。
总体来说,两阶段设计能够克服非随机临床试验的主观性问题,类似于随机临床试验,并获得客观可靠的结论,该方法具有一定的应用价值。
[1] Yue LQ.Statistical and regulatory issues with the application of propensity score analysis to nonrandomized medical device clinical studies.Journal of Biopharmaceutical Statistics,2007,17(1):1-13.
[2] Sedgwick P.What is a non-randomised controlled trial.BMJ,2014:348.
[3] Fairman RM,Criado F,Farber M,et al.Pivotal results of the medtronic vascular talent thoracic stent graft system:the VALOR trial.Journal of vascular surgery,48(3):546-554.
[4] Troisi N,Torsello G,Donas KP,et al.Endurant stent-graft:a 2-year,single-center experience with a new commercially available device for the treatment of abdominal aortic aneurysms.Journal of Endovascular Therapy,17(3):439-448.
[5] Rosenbaum PR,Rubin DB.The central role of the propensity score in observational studies for causal effects.Biometrika,1983,70(1):41-55.
[6] Yue LQ,Lu N,Xu Y.Designing premarket observational comparative studies using existing data as controls:challenges and opportunities.Journal of biopharmaceutical statistics,2014,24(5):994-1010.
[7] Yue LQ,Campbell G,Lu N,et al.Utilizing national and international registries to enhance pre-market medical device regulatory evaluation.Journal of biopharmaceutical statistics,26(6):1136-1145.
[8] Rubin DB.For objective causal inference,design trumps analysis.The Annals of Applied Statistics,2008:808-840.
[9] Rubin DB.Using propensity scores to help design observational studies:application to the tobacco litigation.Health Services and Outcomes Research Methodology,2001,2(3):169-188.
[10]Yue LQ.Regulatory considerations in the design of comparative observational studies using propensity scores.Journal of Biopharmaceutical Statistics,2012,22(6):1272-1279.
[11]Fleming TR,DeMets DL,Roe MT,et al.Data monitoring committees:Promoting best practices to address emerging challenges.Clinical Trials,2017,14(2):115-123.
[12]Krishnankutty B,Bellary S,Kumar NB,et al.Data management in clinical research:an overview.Indian journal of pharmacology,2012,44(2):168.
Two-stageDesigninNon-randomizedMedicalDeviceClinicalTrials
Zhao Yanyan,Xu Yujun,Wang Yang,et al
(StateKeyLaboratoryofCardiovascularDisease,FuwaiHospital,NationalCenterforCardiovascularDiseases,ChineseAcademyofMedicalSciencesandPekingUnionMedicalCollege(100037),Beijing)
ObjectiveThis article introduces the concept,process as well as considerations of two-stage design.The application to non-randomized medical device clinical trials is further illustrated by example of aperipheral vascular stent study.MethodsTwo-stage design based on propensity score serves to make non-randomized clinical trials randomized and prospectively.The details of design process are presented through a non-inferiority trial evaluating the efficacy of a peripheral vascular stent.ResultsTwo-stage design handles the issue of potential biases due to non-randomization and subjective uncertainty when applying propensity score to non-randomized clinical trials,aiming to mimic randomized clinical trials and ensure the objectivity and prospectiveness of both trial design and statistical analysis.ConclusionWith great potential,two-stage design enhances the feasibility of clinical trial and makes it possible to get full use of existing data of high quality,shorten the duration of a trial,improve trial efficiency and draw a reliable conclusion ultimately.
Two-stage design; Propensity score; Medical device; Non-randomized clinical trial
1.中国医学科学院,北京协和医学院,国家心血管病中心,阜外心血管病医院,心血管疾病国家重点实验室(100037) 2.国家心血管病中心
†共同第一作者:赵延延,许毓君
△通信作者:李卫,E-mail:liwei@mrbc-nccd.com
郭海强)
