肺动脉高压中的代谢重编
2017-12-21杨玉霞曹辉林凤越综述姚丽审校
杨玉霞、曹辉、林凤越综述,姚丽审校
综述
肺动脉高压中的代谢重编
杨玉霞、曹辉、林凤越综述,姚丽审校
代谢作为机体基本的生命活动,参与多种生理病理进程,可反映机体实时的病理生理状态。肺动脉高压被称为“心血管病中的癌症”,研究发现代谢重编参与其发病机制。肺动脉高压机体葡萄糖酵解、脂肪酸氧化、氨基酸分解等均发生不同程度的改变,加速了疾病的发展。本文对肺动脉高压病理进程中代谢重编机制作一综述,旨在为这一难治性疾病提供治疗依据。
高血压,肺性;代谢
肺动脉高压(PAH)是继高血压和冠心病的第三类常见心血管病,被称为“心血管病中的癌症”[1],严重危害人类的身心健康,虽然治疗药物不断出现,但其死亡率仍为50%/3年。代谢作为机体基本的生命活动,同时也是机体最终端反应,参与多种疾病进程,Stenmark等提出代谢重编参与PAH的发病机制。本文通过对有关PAH代谢重编的研究作一综述,旨在阐明代谢在PAH中发挥的作用,为PAH的治疗提供依据。
1 PAH及代谢重编概况
PAH是由遗传、缺氧、炎症等因素引起的一种慢性进行性疾病或临床综合征,其诊断标准为静息状态下平均肺动脉压≥25 mmHg(1 mmHg=0.133 kPa)或运动状态下平均肺动脉压≥30 mmHg[2]。PAH基本的病理特征为:肺血管收缩、肺血管重构、原位血栓形成[3-5];其患者临床症状为呼吸困难、运动耐量降低、液体潴留、下肢水肿等[6,7],若不及时治疗2~3年内将死于右心衰竭,因而深入研究PAH的发病机制对发现新的治疗靶点具有指导意义。2012年Fessel等[8]阐明代谢途径异常在PAH发病进程中起关键作用;2015年Stenmark等[9]提出代谢重编参与PAH的发病机制。
代谢重编是指机体在应对内外环境改变的情况下,体内紊乱的代谢途径进行重新编排[10],即重新整合代谢体系,为细胞提供能量[11]。代谢重编既是细胞内网络调控失调的结果,也是维持疾病进程重要的生化基础。正常情况下,代谢维持机体的生长、繁殖以及应对外界环境刺激;疾病时,机体代谢发生显著的异常改变,而异常的代谢又会促进疾病的发展进程,如癌症中代谢改变促进肿瘤细胞的增殖、转移,加速肿瘤的病变过程[12,13]。但疾病时机体并未创造新的代谢反应及途径,只是选择性地激活或抑制其中的代谢通路,改变物质在代谢途径中的流量及流向[14]。那么PAH中代谢重编具体如何发生呢?
2 PAH中的代谢重编
PAH机体代谢变化主要包括葡萄糖酵解、脂肪酸氧化、氨基酸分解,代谢重编的进行加速了疾病的发展进程。因PAH病变主要累及的器官为右心室及肺血管,下面主要对3种营养物质在PAH右心室及肺血管中的代谢变化分别进行阐述。
2.1 葡萄糖酵解
PAH时葡萄糖内稳态失衡,氧化减少,酵解增加,此现象为“Warburg”效应。Warburg效应在PAH发挥关键作用,其可通过沉默交配型信息调节因子2同源蛋白3(Sirt3)/信号传导与转录激活因子(STAT3)/活化T细胞核因子2(NFATc2)、血小板源生长因子(PDGE)/低氧诱导因子-1(HIF-1α)信号通路促进肺动脉平滑肌细胞异常增殖[15];还可通过调节BMPR2基因变异、小窝蛋白-1表达缺乏[16]、HIF-1α活化诱导肺动脉内皮细胞功能障碍,促进PAH的病理发展。因此,可将Warburg效应作为干预PAH的出发点之一。糖酵解增加与糖摄入增加有关,采用正电子发射断层成像(PET)显示与健康对照组相比在PAH患者和动物模型肺和右心室中摄取的18 -氟脱氧葡萄糖(18FDG)明显升高[17]。
PAH时右心室和肺血管中糖分解一致,但诱导因子不同。右心室中的诱导因素是局部缺血,而肺血管中是氧化还原状态调节的活化转录因子,如HIF-1α等[18]。HIF-1α在多种癌细胞中作为Warburg效应的主要调节者[19],在PAH中同样也发挥关键作用。PAH肺血管系统中,线粒体超极化减少超氧歧化酶-2(SOD2)的表达,SOD2的缺乏使氧化还原信号分子过氧化氢产生低于生理水平,进而诱导HIF-1α活化,活化的HIF-1α可诱导丙酮酸脱氢酶激酶(PDK)转录抑制丙酮酸脱氢酶(PDH)[20,21]。PDH的减少抑制了线粒体氧化磷酸化的进行,使线粒体内反应活性氧簇生成减少[22],抑制钾离子通道,使钾离子在细胞内积累引起细胞去极化及胞内钙超载,诱导肺血管收缩,抑制葡萄糖氧化;同时还可诱导活化T细胞核因子(NFAT)活化,促进肺动脉平滑肌细胞增殖抑制其凋亡,诱导肺血管重构,并调节糖分解的酶使葡萄糖氧化减少。HIF-1α和NFAT均能抑制钾离子通道[23]、下调糖代谢酶、抑制糖氧化。PAH机体对糖酵解的依赖增加[18,24],使体内腺嘌呤核苷三磷酸(ATP)生成减少、乳酸产生增加引起酸中毒[20],最终损害右心室功能。
2.2 脂肪酸氧化
不同类型的PAH右心室中脂肪酸氧化程度不同。在肺动脉束扎模型[25]的右心室中脂肪酸氧化增加,如“Randle Cycle”现象(图1),Randle Cycle现象即为葡萄糖与脂肪酸间的竞争关系,增加脂肪酸氧化,抑制葡萄糖氧化,反之亦然[26]。产生相同量的ATP,脂肪酸氧化比葡萄糖氧化多需12%的氧,因此,减少脂肪酸氧化有利于缓解该模型右心室肥厚症状[27]。
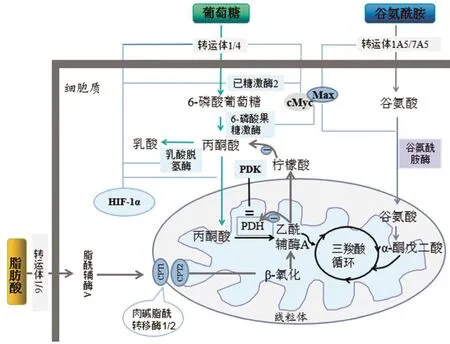
图1 Randle cycle现象和谷氨酰胺分解
在野百合碱等诱导PAH损伤的右心室中脂肪酸转运体CD36及肉碱脂酰转移酶1(CPT1)表达上调引起脂肪酸吸收增加,但脂肪酸氧化减少、消耗减少,从而导致脂肪酸以三酰甘油、神经酰胺等形式聚集在心肌细胞质中引起脂中毒[28],故抑制脂肪酸合成在PAH中起到保护作用[29]。不同PAH模型中脂肪酸氧化程度不同与右心室肥厚状态及代谢酶的表达有关。在血管内皮生长因子拮抗剂Sugen5416合并低氧诱导的PAH模型中酰基辅酶A脱氢酶表达下降引起过氧化物酶体增殖物激活受体γ辅助活化因子1α减少,引起脂肪酸氧化减少;在野百合碱诱导的模型中该酶表达亦减少,而肺动脉束扎模型中该酶表达增加[30]。
脂肪酸氧化在PAH肺血管发病过程中发挥作用[31]。小鼠缺乏降解丙二酰辅酶A的丙二酰辅酶A脱羧酶(MCD),MCD活化的减少能够增加细胞质丙二酰辅酶A的含量、抑制CPT1的表达,从而减少脂肪酸氧化、增加葡萄糖氧化,减缓PAH发展[32]。
鞘磷脂作为脂质的一个分类由鞘氨醇、脂酸和磷酸胆碱构成,在维持细胞膜、信号传递和细胞识别等方面具有重要作用。其中鞘氨醇在鞘氨醇激酶1(SphK1)的作用下生成鞘氨醇-1-磷酸(S1P),S1P作为有效的鞘磷脂中间体在PAH中促进肺血管平滑肌细胞的增殖,引起肺血管重构[33,34]。
2.3 氨基酸分解
谷氨酰胺分解在局部缺血活化cMyc转录途径诱导下被选择性增加[35],水解成氨和谷氨酸。在野百合碱诱导肥厚的右心室中,谷氨酰胺以6倍的速度进行代谢[27,35]。同时,谷氨酰胺分解过程中伴随氮合成代谢增加,进一步促进细胞的生长[36]。
PAH时色氨酸以及精氨酸/一氧化氮代谢程度均发生改变。色氨酸在色氨酸羟化酶作用下生成血清素(5-羟色胺),血清素有利于PAH机体中肺动脉平滑肌细胞增殖,肺血管重构[37]。精氨酸在一氧化氮合酶的作用下生成一氧化氮和瓜氨酸,PAH时体内精氨酸含量减少[38],而精氨酸通过尿素循环产生的鸟氨酸增加,鸟氨酸在鸟氨酸脱羧酶的作用下生成多胺,用于合成脱氧核糖核酸(DNA)和促进细胞生长和分裂。
不同类型PAH的诱因不同(原发性PAH为遗传或基因突变诱导,继发性PAH由缺氧、药物或其他疾病引起),同一类型的PAH在不同组织或器官的诱导因子及病理表现也不同,导致机体不同组织或器官的葡萄糖、脂肪酸、氨基酸等代谢程度或方式存在差异,这种不同的病理表现引起的代谢差异能更清楚地呈现出疾病的发展状态,而局部的代谢重编会引起机体循环物质的变化。代谢物间相互联系相互制约,一个物质的变化连带多种物质的改变,因而异常的代谢可从不同方面促进PAH的病理进程。通过代谢变化分析阐明PAH代谢机制,为PAH治疗提供靶点。
2.4 PAH代谢重编中的代谢抑制剂
代谢酶在代谢通路中发挥关键作用,因而代谢途径中的关键酶/限速酶可以作为干预PAH的靶点。二氯乙酸通过结合在N端区域保守的变构位点抑制所有PDK亚型,它对异常组织有相对特异性而对正常组织鲜有作用[32]。二氯乙酸诱导代谢改变引起线粒体去极化和细胞凋亡,它在多种PAH模型中有利于活化PDH、降低PDH磷酸化,提高葡萄糖氧化与糖酵解的比值(GO/GLY),抑制细胞增殖、促进细胞凋亡、增强血管舒张,逆转肺血管重构[39]。
曲美他嗪和雷诺嗪为部分脂肪酸抑制剂,通过抑制肺血管中脂肪酸氧化过程中的3-酮酰基辅酶A硫解酶,促进葡萄糖经线粒体氧化供能。在肺动脉束扎模型中增加右心室中葡萄糖氧化和ATP水平,改善心输出量和运动耐力[25]。
谷氨酰胺酶抑制剂6-重氮-5-氧代-L正白氨酸(DON)[35]、小分子化合物968[40]和CB-839[41]抑制谷氨酰胺的分解,增加心输出量,减少右心室肥厚,积累PDH活性并增加葡萄糖氧化。上述抑制剂通过抑制代谢途径中的代谢酶,缓解PAH病理进程。
3 PAH代谢重编存在的问题与展望
代谢重编可通过促进肺动脉平滑肌细胞增殖,诱导肺血管重构,加速PAH的发展。但由于PAH发生发展过程中的因素众多以及代谢网络本身的复杂性导致代谢与PAH之间的调控靶点过多,因此如何根据相应的变化给予干预,以及重点干预的靶点还未明确;此外,目前尚无明确的报道确定代谢抑制剂是否可针对性治疗PAH,以及治疗程度如何尚无定论,并且有些抑制剂还未上市,因此建立一对一的代谢调控疾病靶点及药物治疗靶点需进一步深入研究。
代谢组学的出现为代谢研究提供了平台,已有报道通过代谢组学技术分析PAH动物模型及患者中代谢的变化及相应的机制,Lin等[42]通过核磁共振(NMR)技术分析PAH大鼠及药物治疗后代谢模式的变化如脂肪酸β氧化增加,并阐明了其分子机制与糖原合成酶激酶-3(GSK-3)、固醇调节元件结合蛋白1c(SERBP-1c)、肉碱棕榈酰转移酶1(CPT-1)等相关;Zhao等[43]采用液相色谱/气相色谱-质谱联用技术(LC/GC-MS)阐述PAH患者中糖酵解紊乱,脂肪酸代谢物增加等代谢异质化,解释PAH中有特定的代谢途径有利于为血管重构生成ATP。利用代谢组学分析PAH中代谢的变化对发现新的靶点具有指导意义。
综上,代谢重编在PAH病理进程中发挥关键作用。研究PAH中的代谢重编可以从代谢层面深入了解PAH的发病机制,发现PAH新的生物标志物为治疗提供靶点,同时可为探索其他疾病中代谢的变化提供理论依据。
[1] 郑晓东, 朱大岭. 缺氧肺动脉高压发病机制研究进展: 15-LO/15-HETE的作用. 中国药理学通报, 2009, 25: 157-160.
[2] Leopold JA, Maron BA. Molecular mechanisms of pulmonary vascular remodeling in pulmonary arterial hypertension. Int J Mol Sci, 2016, 17: 761.
[3] Bujak R, Mateo J, Blanco I, et al. New Biochemical Insights into the Mechanisms of Pulmonary Arterial Hypertension in Humans. PLoS one, 2016, 11: e0160505.
[4] Sutendra G, Michelakis ED. Pulmonary arterial hypertension:challenges in translational research and a vision for change. Sci Transl Med, 2013, 5: 208.
[5] 郑亚国, 何建国, 熊长明. 肺动脉高压免疫炎症机制研究进展. 中国循环杂志, 2013, 28: 469-471.
[6] Lai YC, Potoka KC, Champion HC, et al. Pulmonary arterial hypertension: the clinical syndrome. Circ Res, 2014, 115: 115-130.
[7] 陈果, 何建国, 柳志红, 等. 不同类型肺动脉高压患者临床特征和血流动力学的比较分析. 中国循环杂志, 2013, 28: 300-303.
[8] Fessel JP, Hamid R, Wittmann BM, et al. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm Circ,2012, 2: 201-213.
[9] Stenmark KR, Tuder RM, El Kasmi KC. Metabolic reprogramming and inflammation act in concert to control vascular remodeling in hypoxic pulmonary hypertension. J Appl Physiol (1985), 2015, 119: 1164-1172.
[10] Peng B, Li H, Peng XX. Functional metabolomics: from biomarker discovery to metabolome reprogramming. Protein Cell, 2015, 6: 628-637.
[11] 黄启超. CD147分子参与肝癌细胞糖代谢重编程的作用及分子调控机制研究 . 第四军医大学, 2013. http: //d. wanfangdata. com. cn/Thesis/D357291
[12] 张亚龙, 房念珍, 尤嘉琮, 等. 肿瘤细胞代谢与肿瘤转移相互关系的研究进展. 中国肺癌杂志, 2014, 29: 812-818.
[13] Benjamin DI, Cravatt BF, Nomura DK. Global profiling strategies for mapping dysregulated metabolic pathways in cancer. Cell Metab, 2012,16: 565-577.
[14] 易梅, 向波, 李小玲, 等. 代谢重编程: 肿瘤的平衡之舞. 中南大学学报(医学版), 2013, 38 : 1177-1187.
[15] Peng H, Xiao Y, Deng X, et al. The warburg effect: a new story in pulmonary arterial hypertension. Clinica Chimica Acta, 2016, 461:53-58.
[16] Zhao YY, Liu Y, Stan RV, et al. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci USA, 2002, 99: 11375-11380.
[17] Marsboom G, Wietholt C, Haney CR, et al. Lung (18)F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am J Respir Crit Care Med, 2012, 185: 670-679.
[18] Archer SL, Fang YH, Ryan JJ, et al. Metabolism and bioenergetics in the right ventricle and pulmonary vasculature in pulmonary hypertension. Pulm Circ, 2013, 3: 144-152.
[19] Semenza GL. Regulation of cancer cell metabolism by hypoxiainducible factor 1. Semin Cancer Biol, 2009, 19: 12.
[20] Rehman J, Archer SL. A proposed mitochondrial-metabolic mechanism for initiation and maintenance of pulmonary arterial hypertension in fawn-hooded rats: the warburg model of pulmonary arterial hypertension. Adv Exp Med Biol, 2010, 661: 171-185.
[21] Archer SL. Acquired mitochondrial abnormalities, including epigenetic inhibition of superoxide dismutase 2, in pulmonary hypertension and cancer: therapeutic implications. Adv Exp Med Biol, 2016, 903: 29-53.
[22] Sutendra G, Dromparis P, Bonnet S, et al. Pyruvate dehydrogenase inhibition by the inflammatory cytokine TNFalpha contributes to the pathogenesis of pulmonary arterial hypertension. J Mol Med (Berl),2011, 89: 771-783.
[23] Michelakis ED, Weir EK. The pathobiology of pulmonary hypertension.Smooth muscle cells and ion channels. Clin Chest Med, 2001, 22: 419.
[24] Sutendra G, Dromparis P, Paulin R, et al. A metabolic remodeling in right ventricular hypertrophy is associated with decreased angiogenesis and a transition from a compensated to a decompensated state in pulmonary hypertension. J Mol Med (Berl), 2013, 91: 1315-1327.
[25] Fang YH, Piao L, Hong Z, et al. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting Randle's cycle. J Mol Med (Berl), 2012, 90: 31-43.
[26] Randle PJ, Garland PB, Hales CN, et al. The glucose fatty-acid cycle.Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet, 1963, 1: 785-789.
[27] Ryan JJ, Archer SL. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res, 2014, 115: 176-188.
[28] Brittain EL, Talati M, Fessel JP, et al. Fatty acid metabolic defects and right ventricular lipotoxicity in human pulmonary arterial hypertension.Circulation, 2016, 133: 1936-1944.
[29] Singh N, Manhas A, Kaur G, et al. Inhibition of fatty acid synthase is protective in pulmonary hypertension. Brit J Pharmacol, 2016, 173:2030-2045.
[30] Gomezarroyo J, Santosmartinez LE, Aranda A, et al. Differences in right ventricular remodeling secondary to pressure overload in patients with pulmonary hypertension. Am J Resp Crit Care, 2014,189: 603-606.
[31] Ryan JJ, Archer SL. Emerging concepts in the molecular basis of pulmonary arterial hypertension: part I: metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation, 2015,131: 1691-1702.
[32] Sutendra G, Bonnet S, Rochefort G, et al. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med, 2010, 2: 44ra58.
[33] Xing XQ, Li YL, Zhang YX, et al. Sphingosine kinase 1/sphingosine 1-phosphate signalling pathway as a potential therapeutic target of pulmonary hypertension. Int J Clin Exp Med, 2015, 8: 11930-11935.
[34] Chen J, Tang H, Sysol JR, et al. The sphingosine kinase 1/sphingosine-1-phosphate pathway in pulmonary arterial hypertension. Am J Respir Crit Care Med, 2014, 190: 1032-1043.
[35] Piao L, Fang YH, Parikh K, et al. Cardiac glutaminolysis: a maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J Mol Med (Berl), 2013, 91: 1185-1197.
[36] Dang CV. Glutaminolysis: supplying carbon or nitrogen or both for cancer cells?. Cell Cycle, 2010, 9: 3884-3886.
[37] Liu Y, Tian H, Yan X, et al. Serotonin inhibits apoptosis of pulmonary artery smooth muscle cells through 5-HT2A receptors involved in the pulmonary artery remodeling of pulmonary artery hypertension. Exp Lung Res, 2013, 39: 70-79.
[38] Zhao YD, Chu L, Lin K, et al. A biochemical approach to understand the pathogenesis of advanced pulmonary arterial hypertension:metabolomic profiles of arginine, sphingosine-1-phosphate, and heme of human lung. PLoS One, 2015, 10: e0134958.
[39] Piao L, Fang YH, Cadete VJ, et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy:resuscitating the hibernating right ventricle. J Mol Med, 2010, 88:47-60.
[40] Xiang Y, Stine ZE, Xia J, et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest,2015, 125: 2293-2306.
[41] Gross MI, Demo SD, Dennison JB, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther, 2014, 13: 890-901.
[42] Lin T, Gu J, Huang C, et al. (1)H NMR-based analysis of serum metabolites in monocrotaline-induced pulmonary arterial hypertensive rats. Dis Markers, 2016, 2016: 5803031.
[43] Zhao Y, Peng J, Lu C, et al. Metabolomic heterogeneity of pulmonary arterial hypertension.PLoS One, 2014, 9:e88727.
黑龙江省教育厅项目(NO.12541421)
150081 黑龙江省哈尔滨市,哈尔滨医科大学药学院 药物化学与天然药物化学教研室
杨玉霞 硕士研究生 主要从事心血管活性药物研究 Email: 2543959867@qq.com 通讯作者:姚丽 Email: 857994401@qq.com
R54
A
1000-3614(2017)12-1246-04
10.3969/j.issn.1000-3614.2017.12.028
2017-06-13)
(编辑:王宝茹)
