利用框内缺失法构建变异链球菌srtA基因缺失株
2017-12-14陈璇刘海霞彭显邹玲
陈璇 刘海霞 彭显 邹玲
口腔疾病研究国家重点实验室 国家口腔疾病临床医学研究中心四川大学华西口腔医院牙体牙髓病科,成都 610041
利用框内缺失法构建变异链球菌srtA基因缺失株
陈璇 刘海霞 彭显 邹玲
口腔疾病研究国家重点实验室 国家口腔疾病临床医学研究中心四川大学华西口腔医院牙体牙髓病科,成都 610041
目的 利用框内缺失法,通过IFDC2基因盒及融合聚合酶链反应(PCR)、同源重组技术构建变异链球菌UA159菌株srtA基因缺失株。方法 PCR扩增变异链球菌UA159菌株srtA基因上下游同源片段及IFDC2基因盒,将这些片段连接、转化入UA159,替换srtA基因同源片段,筛选、鉴定红霉素抗性克隆;PCR扩增、连接UA159 srtA基因上下游同源片段,将连接片段转化入前述红霉素抗性克隆中替换IFDC2同源片段,筛选、鉴定抗苯丙氨酸类似物p-chloro-phenylalanine(p-Cl-Phe)克隆。结果 PCR、琼脂糖凝胶电泳及DNA测序结果均证明srtA基因编码区被完全删除,上下游片段无缝连接,成功构建UA159 srtA基因缺失株。结论 本研究成功构建了无标记的变异链球菌UA159 srtA基因缺失株,为进一步研究该基因在生物膜中的作用及其调控机制奠定了基础。
变异链球菌; srtA基因缺失; 框内缺失法
龋病是发生在牙齿硬组织的慢性感染性疾病[1]。变异链球菌是公认的主要致龋菌之一[1-3],其致龋毒力主要包括:促进细菌在牙齿表面的聚集和黏附、较强的产酸和耐酸能力等[3]。细菌黏附至牙齿表面并形成生物膜是龋病发生的必要条件[1,3]。变异链球菌表面蛋白P1、GpbC及WapA等都与其黏附能力紧密相关[1,4],而这些蛋白都是由变异链球菌srtA基因编码的分选酶sortase A(SrtA)识别并共价锚定到菌细胞壁上发挥作用的[3,5]。变异链球菌srtA基因缺陷可导致其表面蛋白P1等的分布和功能障碍,从而抑制变异链球菌在牙面的黏附增殖和生物膜的形成,其致病能力也相应减弱[1-2,6-7]。因此,SrtA是影响变异链球菌黏附能力的重要毒力因子。
以往研究中使用的srtA基因缺陷株多是采用插入突变和带标记的等位同源突变两种方法构建的,这两种技术中外源DNA片段的引入、明显的极性效应都可能影响研究结果的可信度[8-10]。框内缺失法是目前常用的构建无标记基因缺失株的方法之一[9-10],利用IFDC2基因盒及等位同源交换技术可以高效快捷地完成框内缺失突变株的构建。IFDC2基因盒最初由Xie等[10]设计,由启动子ldh、突变的pheS基因片段(mpheS)及ermAM基因片段三部分构成。本基因盒中的ermAM基因片段使转化子带有红霉素抗性从而实现阳性筛选[10]。pheS基因编码苯丙氨酸-tRNA合成酶α亚基,在其发生突变并整合入受体菌基因组后,可使转化子对苯丙氨酸类似物p-chloro-phenylalanine(p-Cl-Phe)产生获得性敏感[11],利用此原理可以实现转化子的阴性筛选从而获得无标记的基因缺失株。本研究拟利用框内缺失法,通过IFDC2基因盒及相关技术构建无标记的变异链球菌srtA基因缺失株,为进一步研究srtA基因的功能性状及其对变异链球菌致龋毒力的影响提供实验菌株。
1 材料和方法
1.1 菌株及培养条件、试剂及仪器
变异链球菌UA159菌株、IFDC2基因盒由Dr. Justin Merrit(俄克拉荷马大学健康科学中心口腔生物学专业,美国)惠赠。菌株的培养条件为37 ℃、5%CO2兼性厌氧培养。
细菌基因组DNA提取试剂盒[DP302型,天根生化科技(北京)有限公司]、 琼脂糖凝胶DNA回收试剂盒(2500-01型,Omega公司,美国),KODPlus DNA高保真聚合酶试剂盒(KOD-201型,Toyobo公司,日本),琼脂糖(Invitrogen公司,美国),细菌感受态刺激肽(competence stimulating peptide,CSP)(由四川大学口腔疾病研究国家重点实验室提供),红霉素(分装,Amreso公司,美国),p-Cl-Phe(Sigma公司,美国),脑心浸液肉汤(brain and heart infusion,BHI)培养基(Oxoid公司,英国),琼脂(北京博奥拓达科技有限公司)。
细菌培养箱(Anaerobox Ⅳ型,Gene Science公司,美国),超净工作台(1300 Series A2型,Thermo Fisher Scientific公司,美国),聚合酶链反应 (polymerase chain reaction,PCR)仪(S1000 Thermo cycler型)、琼脂糖凝胶电泳仪(Sub-Cell GT Cell型)(Bio-rad公司,美国),分光光度计(Spectronic 200型,Thermo公司,美国)。
本研究所用引物合成、DNA测序均由成都擎科梓熙生物技术有限公司完成。
1.2 细菌培养及细菌基因组DNA(genome DNA,gDNA)提取
将-80 ℃保存的UA159菌株复苏于BHI培养基,形态学和生化鉴定后将平板保存于4 ℃作短期保种,并于平板上挑取单克隆接种于BHI液体培养基中,取过夜培养菌液按细菌基因组DNA提取试剂盒说明提取细菌gDNA,测定纯度及浓度后-20 ℃保存。
1.3 目的片段PCR扩增及连接
以提取所得细菌gDNA及IFDC2基因盒为模板,利用KOD-Plus DNA聚合酶试剂盒PCR扩增UA159 srtA基因上游同源片段(记为up,大小约1 kb)、下游同源片段(记为down,大小约1 kb)、IFDC2片段(记为IFDC2,大小约2 kb);引物根据srtA基因(GenBank accession no NC_004350.2)与IFDC2序列[10]分别设计:upF/upR-IFDC2、ldhF/ermR与dnFIFDC2/dnR(引物序列见表 1),其中引物ldhF和ermR为IFDC2片段的上下游引物;为进行后续融合PCR,引物upR-IFDC2设计为srtA基因上游同源片段的特异性引物upR+ldhF部分反向互补片段,引物dnFIFDC2设计为ermR部分反向互补片段+srtA基因下游同源片段的特异性引物dnF。采用 PCR分别扩增,获得片段up、IFDC2与down。反应条件为94 ℃预变性2 min;94 ℃变性15 s,55 ℃退火30 s,68 ℃分别延伸1 min、2 min 15 s、1 min,35个循环,68 ℃再延伸4 min。4 ℃结束反应后,采用1%琼脂糖凝胶电泳鉴定PCR扩增产物,胶回收并测定产物浓度和纯度,-20 ℃保存。
以up、IFDC2、down三片段为模板,upF和dnR为引物,融合PCR连接上述三片段(记为up-IFDC2-dn,大小约4.1 kb)。反应条件为94 ℃预变性2 min;94 ℃变性15 s,55 ℃退火30 s,68 ℃延伸4 min 15 s,35个循环;68 ℃再延伸4 min。4 ℃结束反应后,采用1%琼脂糖凝胶电泳鉴定 PCR 扩增产物,-20 ℃保存。
1.4 第一步转化及筛选
从1.2中平板上挑取UA159单克隆接种于BHI液体培养基中,取过夜培养菌液按1∶20稀释于BHI培养基后培养2~3 h,直到菌液OD600=0.2~0.3。取菌液按如下分组进行加样,a组:500 µL菌液+5 µL up-IFDC2-dn+0.5 µL CSP(1 µg·µL-1)[12];b组:500 µL菌液+0.5 µL CSP(1 µg·µL-1);c组(阴性对照):500 µL菌液。加样混合后培养2 h,取a、b、c三组菌液各200 µL分别于含有12.5 µg·mL-1红霉素的BHI琼脂平板涂板培养48 h。随机挑取a组平板上部分阳性单克隆接种于含12.5 µg·mL-1红霉素的BHI液体培养基中,以过夜培养菌液及UA159菌液(对照)为模板,加入引物checkF/checkR(引物序列见表1),PCR扩增后1%琼脂糖凝胶电泳检验第一步转化结果,将阳性克隆-80 ℃保种。

表 1 研究所用引物序列Tab1 Primers used in the study
1.5 第二步转化的目的片段PCR扩增及连接
以UA159细菌gDNA为模板,利用KOD-Plus DNA聚合酶试剂盒PCR扩增UA159 srtA基因上游同源片段(记为upΔ,大小约1 kb)及下游同源片段(记为down,大小约1 kb);引物为upF/updnR和dnF/dnR(引物序列见表1),为进行后续融合PCR,引物updnR设计为srtA基因上游同源片段的特异性引物upR+dnF部分反向互补片段。采用PCR分别扩增,获得片段upΔ与down,反应条件94 ℃预变性2 min;94 ℃变性15 s,55 ℃退火30 s,68 ℃延伸1 min,35个循环;68 ℃再延伸4 min。4 ℃结束反应后,采用1%琼脂糖凝胶电泳鉴定PCR扩增产物,胶回收并测定产物浓度和纯度,-20 ℃保存。
以upΔ、down两片段为模板,upF和dnR为引物,融合PCR连接上述两片段(记为upΔ-dn,大小约2 kb)。反应条件94 ℃预变性2 min;94 ℃变性15 s,55 ℃退火30 s,68 ℃延伸2 min 15 s,35个循环;68 ℃再延伸4 min,4 ℃结束反应后,采用1%琼脂糖凝胶电泳鉴定PCR扩增产物,-20 ℃保存。
1.6 第二步转化及筛选
将第一步转化成功的阳性克隆接种至含12.5 µg·mL-1红霉素的BHI液体培养基过夜培养后,于含12.5 µg·mL-1红霉素的BHI琼脂平板上划线培养48 h,挑取单克隆接种于含12.5 µg·mL-1红霉素的BHI液体培养基,取过夜培养菌液按1∶20稀释于不含红霉素的BHI液体培养基培养2~3 h,直至菌液OD600=0.2~0.3。取菌液按如下分组进行加样,a组:500 µL菌液+5 µL upΔ-dn+0.5 µL CSP(1 µg·µL-1);b组:500 µL菌液+0.5 µL CSP(1 µg·µL-1);c组(阴性对照):500 µL菌液;加样混合后培养2 h,取a、b、c三组菌液各200 µL分别于含有4 mg·mL-1p-Cl-Phe的BHI琼脂平板涂板培养48 h。随机挑取a组平板上10个单克隆接种于BHI液体培养基中,以过夜培养菌液及第一步的阳性克隆菌液(对照)为模板,加入引物checkF/checkR(引物序列见表1),PCR扩增后1%琼脂糖凝胶电泳检验第二步转化结果,并将琼脂糖凝胶电泳观察到的阳性克隆送DNA测序,将正确测序结果对应的阳性克隆-80 ℃保种。
2 结果
2.1 红霉素抗性转化子的鉴定
将1.4中a、b、c三组平板培养48 h后,仅a组有菌落生长,b、c组均无菌落长出,表明a组菌落获得红霉素抗性。随机挑取a组部分单克隆培养后用引物checkF/checkR进行PCR,1%琼脂糖凝胶电泳后的结果见图1:以变异链球菌UA159为模板扩增出包含srtA基因的片段长度约为2.6 kb(对照),包含IFDC2的同源片段大小应为4.1 kb,结果均与预期一致,未见非特异性扩增及拖尾现象,说明挑取的所有阳性克隆中srtA基因均被等位同源交换为IFDC2基因盒。
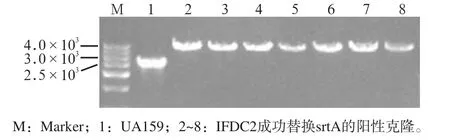
图1 第一次转化后的琼脂糖凝胶电泳检测结果Fig 1 The gel electrophoresis results of the first transformation
2.2 无标记srtA基因缺失株的鉴定
将1.6中a、b、c三组平板培养48 h后,均有菌落长出,a组平板菌落数与b、c组相比无肉眼可见差异。随机挑取a组平板10个单克隆培养以后用引物checkF/checkR进行PCR,1%琼脂糖凝胶电泳后的结果见图2:包含IFDC2基因盒的检测片段长度应为4.1 kb,敲除srtA基因后其上下游无缝连接的片段大小约为2 kb,结果示挑取的10个克隆中有3个其片段大小与预期一致;这3个克隆中有2个其DNA测序结果与已获得的预期序列一致,表明其中srtA基因从起始密码子至终止密码子被完全删除,上下游无缝连接(图3),说明本研究成功构建无标记的变异链球菌UA159菌株srtA基因缺失株。
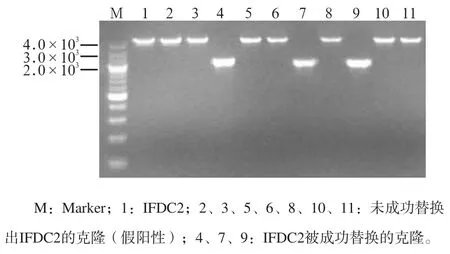
图2 第二次转化后的琼脂糖凝胶电泳检测结果Fig2 The gel electrophoresis result of the second transformation

图3 srtA基因缺失株上下游连接部分测序结果波形图Fig3 The DNA sequencing results at the seam of the constructed srtA-deletion mutant
3 讨论
SrtA是最早从金黄色葡萄球菌中发现的一类重要转肽酶,在革兰阳性菌细胞壁生物合成、生物膜形成及致病能力方面均有重要作用[6,13]。研究[6]发现,srtA基因缺失的金黄色葡萄球菌毒力大幅减低,并且不能在组织中形成脓肿,在放线菌、链球菌、肠球菌、李斯特菌、芽孢杆菌等其他细菌中也有类似发现。在变异链球菌中,SrtA会识别P1、WapA、GbpC、Dex等表面蛋白C末端分选信号中的LPXTG基序,裂解其中苏氨酸-甘氨酸之间的肽键,并将这些蛋白共价连接到菌细胞壁上[1-2,4-5,13-15]。srtA基因缺失会导致上述变异链球菌表面蛋白的分泌、分布及功能异常,影响变异链球菌的初始黏附和生物膜形成,进而减低其致龋性[1-2,6],因此srtA作为变异链球菌重要的毒力基因备受关注。
目前研究变异链球菌基因定义突变的技术中,较成熟的主要有插入突变和带标记的等位同源突变,但插入突变常产生不完全的蛋白产物、用于等位同源替换靶基因的抗性基因片段本身可能携带启动子等因素都可能影响下游基因的表达及细菌表型[8-10]。无标记的基因缺失株构建技术如框内缺失法由于没有引入外源基因标记,很好地规避了对下游基因的极性效应,能更可靠地反映靶基因对转录代谢的影响[9]。Cre/loxP系统、温度敏感质粒或自杀质粒等技术常用来完成无标记缺失株的构建[9-10,12,16],但Cre/loxP系统构建的基因缺失株仍会在细菌基因组中遗留一个loxP位点;温度敏感质粒和自杀质粒的应用则受到质粒种类和靶基因结构等多方面的限制,且上述方法都需要提取质粒并进行扩增,相对耗时较长,还有学者[10]认为在大肠杆菌中引入异种同源DNA片段可能会产生相关毒性。本研究通过设计部分反向互补引物,应用融合PCR构建同源片段,不需要质粒扩增,几小时内就可以获得目的片段,有效地提高了实验效率。
pheS基因在大肠杆菌中编码苯丙氨酸-tRNA合成酶α亚基(PheS),1994年Kast[11]发现点突变的pheS基因(pheS*)编码的产物PheS*在识别苯丙氨酸的同时还能酰基化苯丙氨酸类似物(p-Cl-Phe),其酰化产物会整合入菌细胞蛋白并产生细胞毒性,使pheS*转化子对p-Cl-Phe产生获得性敏感,这个特性随后被应用于突变株的阴性筛选[8,10-11,17]。Xie等[10]在分析变异链球菌PheS蛋白的氨基酸序列后,设计了变异链球菌特异性pheS突变基因——mpheS,并将其与变异链球菌强启动子ldh、红霉素抗性基因片段ermAM相连,构成了IFDC2基因盒,可同时满足阳性和阴性筛选的需求,为框内缺失突变株的构建提供了新方法。相较于Xie等[10]在第二步转化中实现了93%~100%的阳性转化率,本研究中成功率仅为20%,推测原因可能为:本研究涉及的srtA基因表达水平不及Xie等实验中的靶基因nlmA等,在IFDC2基因盒替换srtA基因后,一些调控srtA表达的基因元件仍存在于染色体上,削弱了基因盒中mpheS的表达水平,使其蛋白产物不足以与受体菌正常产生的PheS竞争,一定程度逃避了p-Cl-Phe的细胞毒性,这也解释了为何第二步转化后,含有DNA片段组(a组)平板上菌落数与b、c组无明显差异。综上所述,在运用IFDC2技术构建突变株时,若靶基因表达水平较低或不稳定,在第二步转化后对平板上的阳性转化子进行PCR验证以及DNA测序是必要的,可以避免假阳性结果的产生。
本研究使用融合PCR构建了包含IFDC2基因盒的srtA基因同源片段,在第一次转化中用IFDC2基因盒替换srtA基因,使转化子具有红霉素抗性;然后构建srtA基因上下游无缝连接的同源片段,在第二次转化中替换含有IFDC2基因盒的同源片段,使转化子能够在p-Cl-Phe环境中生长。最后DNA测序证明本研究成功构建了无标记的变异链球菌srtA基因缺失株,其中srtA基因编码序列被完全删除,实现了基因上下游片段的无缝连接,为进一步研究该基因在生物膜中的作用及其调控机制奠定了基础。
[1] Zhuang PL, Yu LX, Tao Y, et al. Effects of missense mutations in sortase A gene on enzyme activity in Streptococcus mutans[J]. BMC Oral Health, 2016, 16:47.
[2] Liao S, Klein MI, Heim KP, et al. Streptococcus mutans extracellular DNA is upregulated during growth in biofilms, actively released via membrane vesicles, and influenced by components of the protein secretion machinery[J]. J Bacteriol, 2014, 196(13):2355-2366.
[3] Lévesque CM, Voronejskaia E, Huang YC, et al. Involvement of sortase anchoring of cell wall proteins in biofilm formation by Streptococcus mutans[J]. Infect Immun, 2005, 73(6):3773-3777.
[4] Li MY, Huang RJ, Zhou XD, et al. Role of sortase in Streptococcus mutans under the effect of nicotine[J]. Int J Oral Sci, 2013, 5(4):206-211.
[5] Lapirattanakul J, Nomura R, Matsumoto-Nakano M, et al. Variation of expression defects in cell surface 190-kDa protein antigen of Streptococcus mutans[J]. Int J Med Microbiol, 2015, 305(3):383-391.
[6] Schneewind O, Missiakas D. Sec-secretion and sortasemediated anchoring of proteins in Gram-positive bacteria[J]. Biochim Biophys Acta, 2014, 1843(8):1687-1697.
[7] Lee SF, Boran TL. Roles of sortase in surface expression of the major protein adhesin P1, saliva-induced aggregation and adherence, and cariogenicity of Streptococcus mutans[J]. Infect Immun, 2003, 71(2):676-681.
[8] Kristich CJ, Chandler JR, Dunny GM. Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis[J]. Plasmid, 2007, 57(2):131-144.
[9] Merritt J, Tsang P, Zheng L, et al. Construction of a counterselection-based in-frame deletion system for genetic studies of Streptococcus mutans[J]. Oral Microbiol Immunol, 2007, 22(2):95-102.
[10] Xie Z, Okinaga T, Qi F, et al. Cloning-independent and counterselectable markerless mutagenesis system in Streptococcus mutans[J]. Appl Environ Microbiol, 2011, 77(22):8025-8033.
[11] Kast P. pKSS—A second-generation general purpose cloning vector for efficient positive selection of recombinant clones[J]. Gene, 1994, 138(1):109-114.
[12] Petersen FC, Scheie AA. Natural transformation of oral streptococci[J]. Methods Mol Biol, 2010, 666:167-180.
[13] Gianella P, Snapp EL, Levy M. An in vitro compartmentalization-based method for the selection of bond-forming enzymes from large libraries[J]. Biotechnol Bioeng, 2016, 113(8):1647-1657.
[14] Marraffini LA, Dedent AC, Schneewind O. Sortases and the art of anchoring proteins to the envelopes of gram-positive bacteria[J]. Microbiol Mol Biol Rev, 2006, 70(1):192-221.[15] Abraham WR. Going beyond the control of quorum-sensing to combat biofilm infections[J]. Antibiotics, 2016, 5(1):3.
[16] Shimshek DR, Kim J, Hübner MR, et al. Codon-improved Cre recombinase (iCre) expression in the mouse[J]. Genesis, 2002, 32(1):19-26.
[17] Zhou C, Shi L, Ye B, et al. pheS*, an effective host-genotypeindependent counter-selectable marker for marker-free chromosome deletion in Bacillus amyloliquefaciens[J]. Appl Microbiol Biotechnol, 2017, 101(1):217-227.
Construction of srtA-deletion mutant of Streptococcus mutans by an in-frame deletion system
Chen Xuan, Liu Haixia, Peng Xian, Zou Ling.
(State Key Laboratory of Oral Diseases amp; Dept. of Conservative Dentistry and Endodontics, National Clinical Research Center for Oral Diseases amp; West China Hospital of Stomatology, Sichuan University, Chengdu 610041, China)
Supported by: The National Natural Science Foundation of China (81570974); The Key Project of the Science and Technology Department of Sichuan Province (2015JY0260). Correspondence: Zou Ling, E-mail: zouling@scu.edu.cn.
Objective To construct srtA-gene deletion mutant of Streptococcus mutans (S. mutans) UA159 with IFDC2 cassette through overlapping polymerase chain reaction (PCR) and allelic homologous recombination. Methods First, the upstream and downstream fragments surrounding the srtA and IFDC2 cassette were PCR amplified and ligated through overlapping PCR. The resulting amplicon was transformed into UA159, and positive transformants were selected on BHI plates containing erythromycin. Second, upstream and downstream fragments of srtA with overlap regions were generated by PCR and were overlapped to create upΔ–down amplicon. Then, the upΔ–down amplicon was transformed into the aforementioned positive transformants and selected on BHI plates containing p-Cl-Phe. Results The PCR analysis and DNA sequencing results indicated that the coding region of the srtA was completely deleted, and the upstream and downstream regions flanking the srtA were ligated seamlessly. Conclusion The markerless srtA-deletion mutant of S. mutans was constructed successfully, which laid a foundation for further study of its biological function and influence on the biofilm formation of S. mutans.
Streptococcus mutans; srtA-deletion mutant; in-frame deletion system
Q 781
A
10.7518/hxkq.2017.06.005
2017-03-28;
2017-05-10
国家自然科学基金(81570974);四川省科技计划项目基金(2015JY0260)
陈璇,硕士,E-mail:252132611@qq.com
邹玲,副教授,博士,E-mail:zouling@scu.edu.cn
(本文编辑 李彩)
