2例不同遗传方式中央轴空病的临床、病理、影像及基因分析☆
2017-12-14林珉婷陈海珠林晓丹何君洁许国荣王柠王志强
林珉婷 陈海珠 林晓丹 何君洁 许国荣 王柠△ 王志强△○☆
·论 著·
2例不同遗传方式中央轴空病的临床、病理、影像及基因分析☆
林珉婷*陈海珠*林晓丹*何君洁*许国荣*王柠*△王志强*△○☆
目的 分析2例不同遗传方式中央轴空病患者的临床、肌肉影像学、病理学及基因突变特点,比较其临床表型和分子遗传学的异同点。方法 详细收集2例患者的临床资料、肌肉MRI及病理学,靶向捕获二代测序进行基因检测,Sanger测序验证及家系共分离分析。结果 2例分别为常染色体隐性(autosomal recessive,AR)和显性(autosomal dominant,AD)患者,表现儿童早期起病,四肢近端无力伴萎缩,面肌受累;双下肢肌肉MRI见广泛肌肉萎缩及脂肪浸润,股直肌回避;病理氧化酶染色见肌纤维典型的轴空结构,AR型存在偏心轴空;发现斯里兰卡肉桂碱受体1(Ryanodine receptor 1,RYR1)基因的3个错义突变,其中一个未报道。结论 本研究的2例经典型患者存在不同遗传方式,在临床表型、受累肌群分布及病理存在诸多异同,可能与RYR1基因的不同突变形式相关,靶向二代测序可以提高确诊率。
中央轴空病 斯里兰卡肉桂碱受体1肌肉磁共振 肌肉病理 靶向捕获二代测序
中央轴空病(central core disease,CCD)是一种罕见的先天性肌病(congenital myopathy,CM),1956年由SHY和MAGEE通过肌肉活检首次报道[1],得名于其特征性肌肉病理改变:线粒体氧化酶类染色见肌纤维中央或偏心、单个或多个、周边境界清楚的轴空结构。CCD以常染色体显性遗传(AD型)为多见,少数为隐性遗传(AR型),根据临床表现的程度和出现时间分为3种类型:轻型、严重型和经典型。轻型为成人期发病,仅在肌肉活检出现肌纤维轴空结构;严重型出现胎儿胎动减弱,甚至胎儿期死亡,出生后吸吮差、呼吸功能不全,运动发育迟缓;经典型表现为婴儿期低肌张力,即松软儿,幼儿期运动发育迟缓。肌肉无力主要分布在近端肢体,一般下肢重于上肢,主要累及骨盆带肌肉和躯干肌,可伴肌肉萎缩、先天性髋关节脱位、脊柱侧弯、骨与关节畸形等并发症。CCD的致病基因 RYR1位于 19q13.1[2],共有 106个外显子,为人类最大的基因之一,直接基因测序难度较大。RYR1编码ryanodine受体1蛋白,一种内质网钙通道蛋白,位于骨骼肌肌质网,组成钙离子释放通道,在骨骼肌运动中发挥兴奋收缩-偶联的作用。国内对于本病的报道较少,以临床和肌肉病理特点的总结为主。我们分析2例基因确诊的经典型CCD,分别是AR型和AD型,并结合文献回顾,以期探讨本病不同病例在临床表型和分子遗传学的诸多异同。
1资料与方法
1.1临床资料 规范采集2例患者的临床资料、体格检查、采用徒手肌力测定法 (manual muscle test,MMT)评肢体肌力。完善心肌酶学谱、肌红蛋白等生化检验,以及心电图、肺功能、肌肉磁共振(MRI,3.0T)和肌电图等辅助检查。收集患者及其家属和100名健康对照的外周静脉血,采用乙二胺四乙酸三钾(EDTA-3K)抗凝。100名健康对照与2个家系均无血缘关系。以上参与人员签署样本采集知情同意书,获得福建医科大学附属第一医院医学伦理委员会伦理支持。
1.2肌肉活检 签署肌肉活检同意书后,行左侧肱二头肌开放式活检标本取材,液氮制备连续冰冻切片,常规进行HE染色、改良Gomori三色染色(MGT)、 三磷酸腺苷酶染色 (ATP PH=4.5;PH=9.6)、油红 O 染色(ORO)、过碘酸盐染色(PAS)、还原型辅酶Ⅰ(NADH-TR),细胞色素C氧化酶染色(COX)、琥珀酸脱氢酶染色(SDH)。
1.3基因变异分析
1.3.1靶向捕获二代测序技术 采用QIAamp DNA Blood Maxi Kit(德国QIGEN公司)抽提基因组DNA。靶向测序涵盖了220种神经肌肉疾病,在线数 据 库 NeuromuscularDisease Center (http://neuromuscular.wustl.edu/time/hmsn.html)提供了基因的基本特征。靶向捕获包含所有的外显子以及外显子上游及下游20 bp内含子中的变异。DNA样 本 由 Nimblegen Exome KitV4 (Roche,Switzerland)捕获后使用 Hiseq3000(美国 illumnia公司)进行二代测序。使用Burrows-Wheeler Aligner (BWA,Version 0.7.12-r1039) 对每一个Read与 hg19基因组参考序列(http://hgdownload.cse.ucsc.edu/)进行比对。二代测序原始数据经过滤和筛选后, 检索数据库 (1000 Genomes,HGMD,Pubmed,UCSC等)确认已知致病突变和单核苷酸多态性,结合患者临床症状,得到候选基因变异位点。 使用 SIFT(http://sift.jcvi.org/)、PolyPhen-2(http://genetics.bwh.harvard.edu/pph2/)和 MutationTaster(http://www.mutationtaster.org)对候选基因变异位点致病性进行预测。
1.3.2 PCR与Sanger测序 采用全血基因组DNA提取试剂盒(天根102DNA抽提试剂盒,中国)提取基因组DNA。针对靶向捕获二代测序筛选的候选基因变异位点应用Sanger测序验证及家系共分离分析。特异性引物设计采用 “Primer Premier 5.0”软件设计,由生工生物工程(上海)股份有限公司合成。25 μL体系进行PCR反应,反应条件:96°C 预变性 5 min,96 °C 变性 30 s,62 °C 退火 30 s,72°C延伸 45 s,共 30个循环,最后 72°C延伸 10 min。扩增产物包含变异位点所在外显子及上下游外显子与内含子交界区。测序结果与Ensemble数据库中参考序列进行比对,验证变异位点。对100名健康对照进行相应位点的检测。
2结果
2.1临床资料 病例1,男,31岁,以“渐进性四肢无力、萎缩约30年”为主诉,自幼运动发育迟缓,适龄不能行走,5岁才能独立行走。体育活动不如同龄儿,跑步动作异常,左右摇摆,蹲立、登梯动作费力,双上肢活动尚好,能举臂、梳头、洗脸、拧毛巾等,伴四肢近端肌肉消瘦。症状进展缓慢,能从事日常生活工作。出生史评分正常,其父、母亲及妹妹均无类似症状(图1A)。查体:双侧眼轮匝肌肌力下降,自然闭眼露白,用力闭眼不紧(图1B),口轮匝肌轻度肌力下降,鼓腮漏气,口唇无肥厚。颈屈肌及颈伸肌肌力5级,双侧三角肌4级,右侧肱二头肌4-级,左侧肱二头肌4级,右侧肱三头肌5-级,左侧肱三头肌4+级,双侧腕伸肌、拇伸肌及指伸肌4级。双侧臀大肌3级,臀中肌3+级,髂腰肌4+级,膕绳肌3+级,股四头肌4级,足背屈肌及趾伸肌4级。肌张力正常,腱反射对称减弱,双侧病理征未引出,无感觉障碍。胸肌稍萎缩,双侧轻度翼状肩,脊柱明显前突伴轻微侧突,伴双侧髋关节轻微畸形,左右摇摆步态。双侧臀部、大腿及小腿肌群存在萎缩,仰卧起坐不能,Beevor’s征阴性。16年前曾于外院行肌肉活检病理染色示非特异性改变,诊断为 “面肩肱型肌营养不良症(FSHD)”。 肌酸激酶(CK)、肌酸激酶同工酶(CKMB),乳酸脱氢酶(LDH)均正常。心电图示正常。超声心动图示三尖瓣区返流。肺功能示:肺活量(VC)为正常值的 72.6%,用力肺活量(FVC)为正常值的75.5%,第一秒最大呼气量(FEV1)为正常值的84.6%,为轻度限制性肺通气功能障碍。肌电图示:四肢肌肉存在肌源性损害;瞬目反射波幅低;双侧面神经运动CMAP波幅低;左口轮匝肌运动单位时限窄。
病例2,女,25岁,以“渐进性四肢无力约 20年”为主诉,自幼运动能力较同龄人为差,跑步、登梯、跳跃等动作缓慢,易摔倒,能参加体育锻炼。症状缓慢进展,现蹲立、登梯、爬山等感双下肢易疲乏,以右下肢无力较左侧明显,伴下肢近端肌肉萎缩。双上肢活动能力稍差,无抬举、提重物困难,平卧位时需翻身侧撑起身,能胜任日常生活工作。出生史正常,无其他病史。其父亲52岁,从小开始有类似病史,目前以双下肢无力和萎缩为明显,能自己行走、蹲立(图2A)。查体:双侧眼轮匝肌力稍下降,闭眼无露白,眼轮匝肌闭合力弱。(图2B),鼓腮无漏气。颈屈肌2级,颈伸肌5级,三角肌4级,肱二头肌及肱三头肌肌力4+级,髂腰肌4-级,膕绳肌4+级,臀中肌4+,臀大肌4-级,四肢远端肌力 5级。四肢肌张力正常,腱反射对称迟钝。右下肢较左下肢明显消瘦、萎缩。步态未见异常,无脊柱侧弯及髋关节脱位等。 CK:50 U/L (30~135 U/L),CKMB:6 U/L (<25 U/L),AST:18 (0 ~46) U/L,LDH:177(109~245) U/L,肌红蛋白 MYO:34.21(0~110)μg/L。心电图及超声心动图示正常。肺功能示:肺活量(VC)为正常值的46.8%,用力肺活量(FVC)为正常值的40.0%,第1秒最大呼气量(FEV1)为正常值的 44.5%,最大呼气流量(PEF)为正常值的42.6%,为重度限制性肺通气功能障碍。肌电图示肌源性损害。
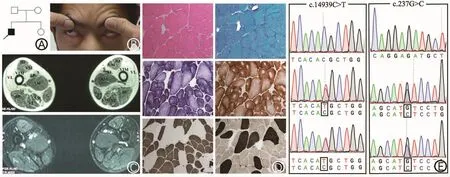
图1 病例1的家系图、面部体征、下肢肌肉MRI及病理结果。 A:家系图;B:双侧眼轮匝肌无力;C:大腿肌肉MRI,股中间肌及股外侧肌严重受累,而股直肌回避;(RF:股直肌;VL:股外侧肌;VIM:股中间肌;VM:股内侧肌;AL:长收肌;S:缝匠肌;AM:大收肌;G:股薄肌;SM:半膜肌;ST:半腱肌;BF:股二头肌)D:肌肉病理染色:HE染色见肥大和萎缩纤维散在分布,MGT染色未见肌纤维内紫红色物质沉积,NADH-TR染色及 COX和SDH复合染色见肌纤维中央轴空结构,部分为偏心轴空,ATP染色见Ⅰ型纤维占优势;E:RYR1基因Sanger测序图示正常序列和患者c.14939C>T和c.237G>C的复合杂合突变,分别来源于其父亲和母亲。
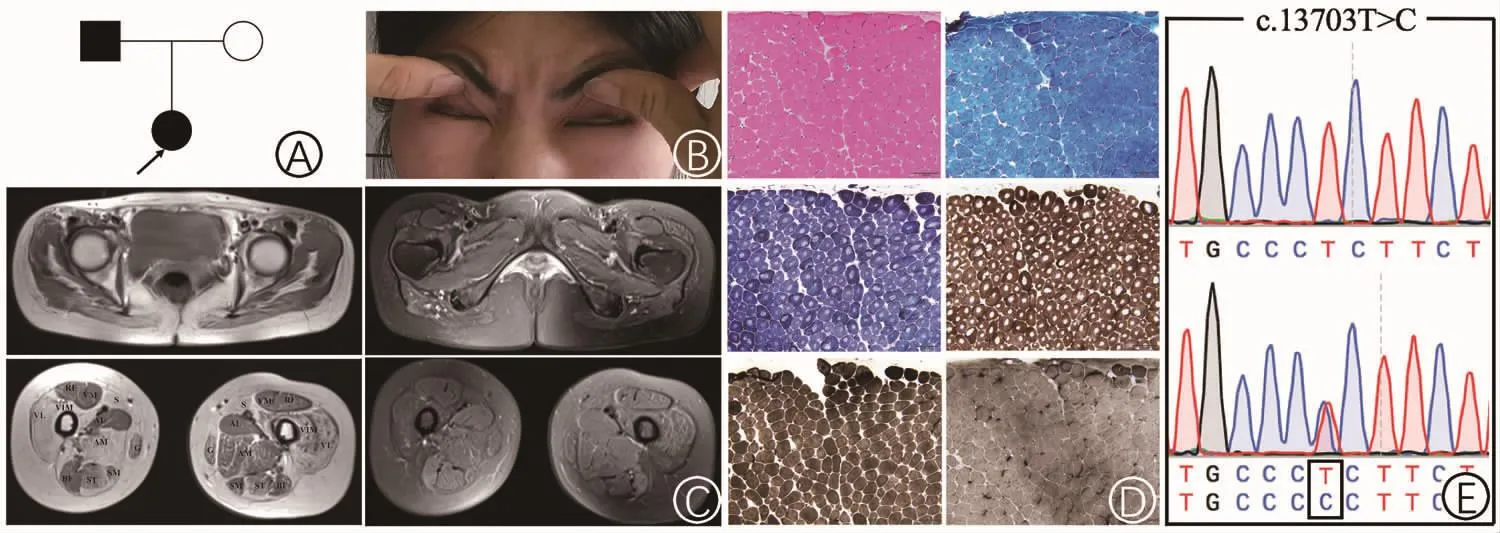
图2 示病例2的家系图、面部体征、骨盆及双下肢肌肉MRI和病理结果。A:家系图;B:双眼轮匝肌无力;C:骨盆及大腿肌肉磁共振,双侧臀大肌及右侧大腿股中间肌、股外侧肌群、股二头肌、大收肌,缝匠肌均匀高信号,左侧大腿股外侧肌、股中间肌、股二头肌高信号,双侧不对称受累;(RF:股直肌;VL:股外侧肌;VIM:股中间肌;VM:股内侧肌;AL:长收肌;S:缝匠肌;AM:大收肌;G:股薄肌;SM:半膜肌;ST:半腱肌;BF:股二头肌)D:肌肉病理染色:HE染色可见小圆形肌纤维散在分布,MGT染色未见肌纤维内紫红色物质沉积,NADH-TR染色及COX和SDH复合染色见全部肌纤维中央轴空结构,ATP染色见Ⅰ型纤维占绝对优势;E:RYR1基因Sanger测序图示正常序列和患者c.13703T>C杂合突变。
2.2大腿肌肉MRI病例1见股外侧肌、股中间肌、股内侧肌、缝匠肌、大收肌在T1相呈均匀高信号,半膜肌呈混杂信号;脂肪抑制相见受累肌肉内脂肪信号被抑制(图1C)。病例2见双侧臀大肌及右侧大腿股中间肌、股外侧肌群、股二头肌、大收肌、缝匠肌T1相均匀高信号,半腱肌、半膜肌、股薄肌混杂信号;左侧大腿股外侧肌、股中间肌、股二头肌高信号,半腱肌、半膜肌、股薄肌、大收肌混杂信号;脂肪抑制相见受累肌肉内脂肪信号被抑制(图 2C)。
2.3骨骼肌病理 病例1左侧肱二头肌 (图1D)HE染色可见肌纤维肥大与萎缩,直径大小不一,未见肌纤维坏死与再生,未见肌纤维分裂及核内移。HE及MGT染色可见肌束内结缔组织增生,肌间小血管壁无增厚,未见异常物质沉积,血管周围未见炎细胞浸润。ATP酶染色(PH=4.5/9.4)见Ⅰ型肌纤维相对占优势,Ⅱ型肌纤维明显肥大。在磷酸化酶及氧化酶染色(NADH-TR及COX+SDH复合染色)见Ⅰ型肌纤维中央位置出现单个、周边境界清楚的轴空结构,部分肌纤维呈偏心轴空改变,Ⅱ型肌纤维未见轴空结构。ACP、ORO及PAS染色未见明显异常。
病例2左侧肱二头肌(图2D)HE染色见轻度萎缩的肌纤维散在分布,未见明显肥大,未见劈裂纤维及内核纤维。MGT染色未见异常物质沉积,未见破碎红纤维。ATP酶染色(PH=4.5/9.4)见Ⅰ型肌纤维占绝对优势,几乎未见Ⅱ型肌纤维。NADH-TR及COX+SDH复合染色可见Ⅰ型肌纤维中央单个、周边境界清楚的淡染轴空结构。ACP、ORO及PAS染色未见明显异常。
2.4基因检测 靶向捕获二代测序包囊括了220种神经肌肉疾病,包含180个致病基因。97.24%目标碱基测序深度达到50X,99.17%碱基测序深度达到30X。数据经过滤和筛选后,发现病例1为RYR1基因的3号外显子c.237G>C (p.E79D)和103号外显子c.14939C>T(p.T4975M)的复合杂合突变(图1E);其中c.237G>C为新发变异,未见文献报道,c.14939C>T为已报道致病突变[3]。经Sanger测序和家系共分离分析证实c.237G>C来源于母亲,c.14939C>T来源于父亲。对于新发变异c.237G>C的致病性进行多项验证:在100个健康对照人群的200条染色体中未发现该变异;软件预测SIFT为致病(Score=0),PolyPhen-2为可能致 病 (Score=0.861),MutationTaster 为 致 病 ,在ExAC和1000G数据库中尚未见报道;在不同物种中为高度保守序列。病例 2为c.13703T>C(p.L4568P)杂合突变(图 2E),为已报道致病突变[4]。病例2的父母亲外周静脉血未提供,未进行家系共分离分析。
3讨论
中央轴空病作为CM的常见类型,具有一些共同的临床表现:起病早,全身性肌无力和肌张力下降,运动发育迟缓,可伴有骨骼发育异常等。这些表现缺乏特异性,且具有较高的临床异质性。CCD多为常染色体显性遗传,近年有少数的常染色体隐性病例报道,未见两者有何临床差异的总结。在本研究中,病例1为AR型,病例2为AD型,两者均表现为早期起病的双下肢近端无力和萎缩,进展缓慢,病情温和,属于经典型表现;同时,面肌轻度受累导致闭睑乏力,肩胛带肌萎缩导致轻度翼状肩,曾诊断为 “面肩肱型肌营养不良症”,后者有AD遗传家族史,但多数青少年期不对称起病,以上肢体肌无力为首发,伴“Beevor’s征”。此外,还需与肢带型肌营养不良症相鉴别。两者病程中均无高热病史,心肌酶学水平正常,无心脏受累的表现。有报道部分患者出现呼吸功能不全[5],病例1和病例2分别存在轻度和重度限制性肺通气功能障碍,但无明显临床症状,可能与呼吸肌容易累及有关。2例患者除了性别之外,也存其他临床差异,病例1合并髋关节轻微畸形、脊柱侧弯,步态异常突出;病例2未见骨关节异常,步态尚可,临床表型更轻。
肌肉MRI作为评估肌群受累程度和分布模式的重要手段,在不同类型肌病有相应变化规律,有助于鉴别诊断[6-7]。CCD肌肉MRI具有选择性肌群受累,表现为大腿股内侧肌、股外侧肌、股中间肌、缝匠肌、大收肌受累,而股直肌、长收肌、半腱肌、股薄肌保留;小腿比目鱼肌和腓肠肌内侧头明显受累,而胫前肌和腓肠肌外侧头保留[8-11]。本研究2例患者大腿肌肉MRI除典型肌肉受累外,亦见半腱肌、半膜肌、股薄肌不同程度的受累,股直肌保留。肌肉MRI与肌力评估结果基本一致。FISHER等[12]报道AD型CCD有臀大肌受累,病例2(AD型CCD)见臀大肌对称受累,但双侧下肢肌肉不对称受累,而病例1(AR型CCD)患者双侧下肢肌肉对称性脂肪浸润。CCD患者肌肉选择性受累的机制尚不明确。
先天性肌病具有相似的临床表现,常规实验室检查缺乏特异性,肌肉病理活检可以直接获得有价值的诊断依据。典型的CCD肌肉病理为线粒体氧化酶类染色见Ⅰ型肌纤维中央、单个、周边境界清楚的轴空结构,轴空样结构贯穿全长肌纤维;ATP染色见Ⅰ型肌纤维占优势,Ⅱ型肌纤维减少。电镜下见线粒体的缺失、Z线波浪式结构混乱、灶性肌小节断裂[13]。病例1的Ⅰ型肌纤维上存在典型中央轴空,部分肌纤维中见偏心轴空。其在10多年前于外院行肌肉活检未见典型中央轴空,提示 CCD肌肉病理改变可能随病程进展而发生变化[11]。病例2的肌肉病理为典型的中央轴空结构弥漫分布在Ⅰ型纤维,几乎未见Ⅱ型肌纤维,与文献报道CCD病理特点一致。
RYR1基因突变集中在3个热点区:热点区1(外显子 1~17),热点区 2(外显子 39~46)和热点区 3(外显子 90~104)[14-15]。热点区 3 编码 RYR1 蛋白的C端,该区突变可出现CCD的典型临床症状,被认为与CCD密切相关。热点区1编码RYR1蛋白的N端,该区突变除了导致CCD,还可能与恶性高热(malignant hyperthermia,MH)相关[16]。 恶性高热是一种药源性疾病,手术麻醉使用吸入麻醉剂或去极化肌肉松弛剂可诱导易感个体出现急性高代谢症候群,包括高热、肌强直、心动过速、心律失常、酸中毒和横纹肌溶解等。基于对热点区的认识,既往研究者通过筛查RYR1基因的90~106号外显子查找突变位点[5,17-18],仅筛查热点区3可能遗漏其他外显子的突变位点。本研究的2例病人均采用靶向二代测序手段对相关的肌肉疾病致病基因进行无偏倚筛查。病例1复合杂合突变c.237G>C位于 RYR1蛋白的 N端,c.14939C>T位于RYR1蛋白的C端,分别来源于无症状双亲,其临床表型亦符合CCD的典型症状,而N端突变可能导致MH,在CCD患者行手术麻醉前应警惕有恶性高热发生的可能。WU等[4]报道肌肉病理特点与基因突变位点相关,其病理上存在中央轴空和偏心轴空并存的结构改变,可能是RYR1基因C端突变位点与N端突变共同作用结果,病例1病理亦表现为中央轴空和偏心轴空并存,与文献报道相一致。病例2的基因改变c.13703T>C位于RYR 1蛋白的C端,病理表现为肌纤维中央、单一的轴空结构,为典型的CCD病理表现。此外,RYR1基因突变和多微轴空病相关[19-20],肌肉病理染色与RYR1基因检测相结合有利于CCD的明确诊断。提示CCD表型轻重及遗传方式与RYR1基因突变位点不同影响蛋白功能区不同相关。
本研究报道的2例经典型CCD患者存在不同遗传方式,在临床表型、受累肌群分布及病理特点存在诸多异同,可能与RYR1基因的不同突变形式相关,通过靶向二代测序可以提高确诊率。
致谢:感谢福建医科大学病理学系张文敏教授在肌肉病理诊断上的协助。
[1]MAGEE KR,SHY GM.A new congenital non-progressive myopathy[J].Brain,1956,79(4):610-621.
[2]MULLEY JC,KOZMAN HM,PHILLIPS HA,et al.Refined genetic localization for central core disease [J].Am J Hum Genet,1993,52(2):398-405.
[3]KLEI A,HEINZ J,EMMA C,et al.Muscle Magnetic Resonance Imaging in Congenital Myopathies Due to Ryanodine Receptor Type 1 Gene Mutations[J].Arch Neurol,2011,68(9):1171-1179.
[4]WU S,IBARRA MC,MALICDAN MC,et al.Central core disease is due to RYR1 mutations in more than 90%of patients[J].Brain,2006,129(Pt 6):1470-1480.
[5]吴士文,马维娅,于生元,等.28例中央轴空病的临床病理分析[J].中华神经医学杂志,2006,(6):597-600.
[6]LIU XY,JIN M,WANG ZQ,et al.Skeletal Muscle Magnetic Resonance Imaging of the Lower Limbs in Late-onset Lipid Storage Myopathy with Electron Transfer Flavoprotein Dehydrogenase Gene Mutations[J].Chin Med J (Engl),2016,129(12):1425-1431.
[7]CASTIGLIONI C,CASSANDRINI D,FATTORI F,et al.Muscle magnetic resonance imaging and histopathology in ACTA1-related congenital nemaline myopathy [J].MuscleNerve,2014,50(6):1011-1016.
[8]JUNGBLUTH H,DAVIS MR,M u..LLER C,et al.Magnetic resonance imaging of muscle in congenital myopathies associated with RYR1 mutations[J].Neuromuscul Disord,2004,14(12):785-790.
[9]ZHOU H,JUNGBLUTH H,SEWRY CA,et al.Molecular mechanismsand phenotypic variation in RYR1-related congenital myopathies[J].Brain,2007,130(Pt 8):2024-2036.
[10]KLEIN A,JUNGBLUTH H,CLEMENT E,et al.Muscle Magnetic Resonance Imaging in Congenital Myopathies Due to Ryanodine Receptor Type 1 Gene Mutations [J].Arch Neurol,2011,68(9):1171-1179.
[11]JUNGBLUTH H,M?LLER CR,HALLIGER-KELLER B,et al.Autosomalrecessive inheritance of RYR1 mutationsin a congenital myopathy with cores [J].Neurology,2002,59 (2):284-287.
[12]FISCHER D,HERASSE M,FERREIRO A,et al.Muscle imaging in dominant core myopathies linked or unlinked to the ryanodine receptor 1 gene[J].Neurology,2006,67(12):2217-2220.
[13]FISCHER D,HERASSE M,FERREIRO A,et al.Muscle imaging in dominant core myopathies linked or unlinked to the ryanodine receptor 1 gene[J].Neurology,2006,67(12):2217-2220.
[14]ZHOU H,JUNGBLUTH H,SEWRY CA,et al.Molecular mechanisms and phenotypic variation in RYR1-related congenital myopathies[J].Brain,2007,130(Pt 8):2024-2036.
[15]DAVIS MR,HAAN E,JUNGBLUTH H,et al.Principal mutation hotspot for central core disease and related myopathies in the C-terminal transmembrane region of the RYR1 gene[J].Neuromuscul Disord,2003,13(2):151-157.
[16]ROBINSON R,CARPENTER D,SHAW MA,et a1.Mutations in RYRl in malignant hyperthermia and central core disease[J].Hum Mutat,2006,27(10):977-989.
[17]GU M,ZHANG S,HU J,et al.Novel RYR1 missense mutations in six Chinese patients with central core disease [J].Neurosci Lett,2014,566:32-35.
[18]CHANG X,JIN Y,ZHAO H,et al.Clinical features and ryanodine receptor type 1 gene mutation analysis in a Chinese family with central core disease [J].J Child Neurol,2013,28(3):384-388.
[19]JUNGBLUTH H,ZHOU H,HARTLEY L,et al.Minicore myopathy with ophthalmoplegia caused by mutations in the ryanodine receptor type 1 gene [J].Neurology,2005,27,65(12):1930-1935.
[20]MONNIER N,FERREIRO A,MARTY I,et al.A homozygous splicing mutation causing a depletion of skeletal muscle RYR1 is associated with multi-minicore disease congenital myopathy with ophthalmoplegia [J].Hum Mol Genet,2003,12 (10):1171-1178.
Clinical,pathological,imaging and genetic analysis of two cases of central core disease with different inheritance modes.
LIN Minting,CHEN Haizhu,LIN Xiaodan,HE Junjie,XU Guorong,WANG Ning,WANG Zhiqiang.Department of Neurology and Institute of Neurology,First Affiliated Hospital,Fujian Medical University,Fuzhou,China.Fujian Key Laboratory of Molecular Neurology,Fuzhou 350005.Tel:0591-87982772.
Objective To study the clinical,pathological,imaging features of two cases of central core disease(CCD)with different inheritance and to explore the similarities and differences between autosomal recessive CCD (ARCCD)and autosomal dominant CCD (AD-CCD).MethodsClinical manifestations,family history,muscle MRI and muscle biopsy were collected.Targeted next generation sequencing(NGS)and sanger sequencing were applied for genetic analysis.Co-segregation analysis was further conducted in one family.ResultsTheir common clinical manifestations included childhood early-onset proximal limbs muscle weakness and dystrophy accompanied with facial involvement.The MRI revealed extensive muscular dystrophy and fatty filtration in the both thighs,but not in rectus femoris.Pathology of skeletal muscle showed typical central cores in typeⅠmuscle fibers and eccentric cores only in AR-CCD.Targeted NGS identified 3 missense mutations in RYR1,including one novel mutation. Conclusion The present study has described clinical and pathological features of two typical CCD patients with different inheritance,which may be associated with the different mutations in RYR1 gene.Targeted NGS apparently improves the genetic diagnosis of CCD.
Central core disease Ryanodine receptor 1 Muscle MRIMuscle pathologicalTargeted next generation sequencing
10.3969/j.issn.1002-0152.2017.09.001
☆ 国家自然科学基金资助项目(编号:81671237);国家自然科学基金资助项目(编号:U1505222);福建省科技创新联合资金资助项目(编号:2016Y9010);福建省自然科学基金资助项目(编号:2017J01196)
* 福建医科大学附属第一医院神经内科(福州 350005)
△ 福建省神经分子重点实验室
○☆ 通信作者(E-mail:fmuwzq@fjmu.edu.cn)
R730.264
A
2017-07-05)
李立)
