灰葡萄孢BcKMO基因的原核表达分析
2017-12-12刘媛媛姜婷婷时翠平邢继红董金皋
王 敏,刘媛媛,周 帆,姜婷婷,郑 旭,张 靖,时翠平,邢继红,董金皋
(河北农业大学,真菌毒素与植物分子病理学实验室,河北 保定 071001)
灰葡萄孢(Botrytiscinerea)是一种死体营养型植物病原真菌,其寄主范围广泛,可引起蔬菜、水果等200多种植物的灰霉病,给农业生产造成巨大经济损失[1-3]。目前,灰霉病预防以化学药剂为主,但其连续使用,易使病菌产生抗药性,且危害人畜安全,对环境造成压力,严重破坏生态平衡[4-5]。因此,寻找一种快速抑制病原菌、特异性强、对环境友好的新型生物杀菌剂迫在眉睫[6]。犬尿氨酸单加氧酶(Kynurenine 3-monooxygenase,KMO)是NADPH依赖的黄素蛋白羟化酶,位于犬尿氨酸途径的中心。KMO催化犬尿氨酸在它的酚环第3位羟基化,生成3-羟基犬尿氨酸(3-HK)[7-9]。犬尿氨酸途径主要存在于色氨酸分解代谢,广泛存在于真核生物和原核生物中。犬尿氨酸代谢产生一些生物活性中间体,带有不同生理行为,参与各种神经退行性疾病的发病机理,包括阿尔茨海默病、帕金森症和亨廷顿疾病[10-11]。近年来,在一些微生物中也发现了类似的犬尿氨酸途径,如荧光假单胞菌(Pseudomonasfluorescens)和哈氏噬纤维菌(Cytophagahutchinsonii)[12-14]、绿脓单胞菌(Pseudomonasaeruginosa)[15]、金羊毛链霉菌(Streptomyceschrysomallus)[16],犬尿氨酸途径在安曲霉素生物合成中起重要作用[17]。由此,笔者推测灰葡萄孢中也存在犬尿氨酸途径[18]。河北农业大学真菌毒素与植物分子病理学实验室前期研究中,克隆了灰葡萄孢BcKMO基因,明确了BcKMO正调控灰葡萄孢的生长、发育,负调控灰葡萄孢的致病力[19];确定了BcKMO通过调控灰葡萄孢的胞壁降解酶活性、毒素活性、产酸能力及致病相关基因的表达而影响病菌的致病力[20]。但是BcKMO基因编码产物的功能及其与病菌致病力调控之间的关系尚未明确。因此,本试验对灰葡萄孢BcKMO基因进行原核表达分析,获得纯化的BcKMO蛋白,为下一步的酶活力测定及其互作蛋白的筛选奠定基础,并为阐明单加氧酶及其所在的犬尿氨酸途径与病菌致病力之间的关系提供理论依据。
1 材料和方法
1.1 试验材料
灰葡萄孢野生型菌株BC22由河北农业大学真菌毒素与植物分子病理学实验室保存;大肠杆菌DH5α和BL21、限制性内切酶、T4 DNA连接酶购自TaKaRa公司;pMD19-T载体、pGEX4T-1载体购自Pharmacia Biotech公司;RNA提取试剂盒、反转录试剂盒购自北京全式金公司。
1.2 试验方法
1.2.1 引物设计 根据BcKMO基因序列,采用Primer 5.0软件设计两端带有NotⅠ和SmaⅠ酶切位点的特异性引物yhKMO-F(5′-ATAAGAATGCGG CCGCATGCCGTCCTTGTTGATC-3′)和yhKMO-R(5′-TCCCCCGGGAATGTCTGCACCATCCTCTAAAG-3′)。其中下划线的部分分别是NotⅠ和SmaⅠ的酶切位点。
1.2.2BcKMO基因的克隆 以灰葡萄孢野生型菌株BC22为试材,提取RNA,反转录成cDNA,用高保真Taq酶扩增BcKMO基因。PCR反应体系:模板cDNA 2 μL、yhKMO-F(10 μmol/L) 0.5 μL、yhKMO-R(10 μmol/L) 0.5 μL、LATaq酶0.5 μL、10×LA PCR Buffer Ⅱ 2.5 μL、10 mmol/L dNTP 2 μL、ddH2O 18 μL,共25 μL。经PCR扩增,琼脂糖凝胶电泳检测,目的条带回收后与载体pMD19连接,连接后的产物转化大肠杆菌DH5α感受态细胞,阳性克隆测序正确后进行质粒pMD19-T-BcKMO的提取。
1.2.3 原核表达载体的构建与鉴定 用NotⅠ和SmaⅠ分别对pMD19-T-BcKMO和pGEX4T-1质粒进行双酶切,回收目的片段后连接,转化大肠杆菌DH5α感受态细胞,通过菌落PCR、酶切验证及测序验证后,确定原核表达载体pGEX4T-1-BcKMO-GST构建成功。将pGEX4T-1-BcKMO-GST和pGEX4T-1分别转化大肠杆菌BL21,确定阳性克隆进行蛋白的诱导表达。
1.2.4 不同浓度的IPTG诱导对BcKMO蛋白表达的影响 将转化了pGEX4T-1-BcKMO-GST和pGEX4T-1的大肠杆菌BL21分别在LB培养基中培养8 h,37 ℃转接,培养至OD600约为0.7,分别加入不同浓度的IPTG至终浓度为0.1,0.2,0.4,0.8,1.0 mmol/L,28 ℃诱导14 h,收集菌体,弃上清,用pH值8.8 Tris-HCl和5×SDS-PAGE Loading Buffer重悬沉淀,沸水浴10 min,离心取上清进行SDS-PAGE,检测目的蛋白的表达情况,进而确定最适IPTG诱导浓度。
1.2.5 不同诱导时间对BcKMO蛋白表达的影响 利用上述得到的最适IPTG诱导浓度,28 ℃诱导pGEX4T-1-BcKMO-GST和pGEX4T-1的表达,诱导时间分别为4,8,12,16,20 h,收集菌体,提取蛋白,进行SDS-PAGE,确定诱导表达的最佳时间。
1.2.6 BcKMO蛋白的纯化 利用上述得到的最适IPTG诱导浓度和最佳诱导时间,在28 ℃进行诱导表达,收集菌体,弃上清,用pH值8.8 Tris-HCl重悬菌体沉淀,加入蛋白酶抑制剂,利用超声细胞破碎仪破碎菌体,超声功率100 W,工作10 s,间歇20 s,循环30次,离心收集上清,4 ℃保存备用。
使用ProteinIsoTMGST Resin对收集的上清进行目的蛋白的纯化,取1 mL GST beads加入20 mL Binding Buffer (50 mmol/L Tris-HCl pH值8.0,150 mmol/L NaCl)进行平衡,4 ℃混匀10 min,700 r/min离心5 min,弃上清,加入20 mL Binding Buffer重悬,重复洗3次,弃上清,冰上备用;将菌体上清加入上述平衡的GST beads,4 ℃混匀2 h,700 r/min离心5 min,弃上清,留沉淀;向沉淀中加入20 mL Binding Buffer(含0.1% TritonX-100),4 ℃混匀10 min,700 r/min离心5 min,弃上清,重复洗涤2次,弃上清,留沉淀;向沉淀中加入500 μL洗脱缓冲液(50 mmol/L Tris-HCl pH值8.0,10 mmol/L 谷胱甘肽),4 ℃混匀1 h,700 r/min离心5 min,收集洗脱液,重复洗2次,分别得到洗脱液,进行SDS-PAGE,检测目的蛋白的纯化情况。
1.2.7 Western Blot分析 利用Western Blot技术,对目的蛋白进行检测。分别将纯化后的蛋白和菌体的全蛋白进行SDS-PAGE,利用转膜仪转到PVDF膜上,用5%脱脂奶粉封闭2 h,用PBST洗涤4次,按1∶1 000加入GST Tag Antibody,杂交1 h,用PBST洗涤4次,每次5 min,按1∶10 000加入goat anti-mouse HRP antibody,杂交1 h,用PBST洗涤5次,每次5 min,然后在PVDF膜上加入Western Blot发光检测液A、B混合液1 mL,用保鲜膜封好,放在暗匣内,在暗室进行显影[21]。
2 结果与分析
2.1 BcKMO基因原核表达载体的构建
提取灰葡萄孢野生型菌株BC22总RNA,反转录成cDNA,用高保真Taq酶扩增BcKMO基因(图1-A)。回收PCR扩增产物,与pMD19-T载体连接,转化大肠杆菌,得到pMD19-T-BcKMO阳性克隆。用NotⅠ和SmaⅠ进行酶切检测,获得了2条与目的片段大小一致的条带(图1-B),表明BcKMO基因已连接pMD19-T载体。将pGEX4T-1载体和测序正确的pMD19-T-BcKMO载体分别进行NotⅠ和SmaⅠ双酶切,回收目的片段,用T4连接酶连接,转化大肠杆菌,通过酶切验证获得了与目的条带大小一致的条带(图1-C),表明原核表达载体pGEX4T-1-BcKMO-GST构建成功。
2.2 不同浓度的IPTG诱导对BcKMO蛋白表达的影响
为确定IPTG诱导使用的最适浓度,37 ℃转接培养至OD600约为0.7,分别加入不同浓度的IPTG至终浓度为0.1,0.2,0.4,0.8,1.0 mmol/L,28 ℃诱导14 h,收集菌体,以未经诱导的菌体为对照,进行SDS-PAGE检测。结果发现,转化pGEX4T-1载体的菌株经诱导表达出约26 kDa的蛋白条带,与GST蛋白大小一致(图2-A),而带有pGEX4T-1-BcKMO-GST载体的菌株诱导表达出约71 kDa的蛋白条带(图2-B),与融合蛋白大小一致(GST蛋白为26 kDa,BcKMO蛋白45 kDa)。当IPTG浓度为0.1 mmol/L时,融合蛋白的表达水平较低;当IPTG浓度为0.2~1.0 mmol/L时,融合蛋白的表达水平没有明显变化(图2-B),由此确定IPTG诱导使用的最适浓度为0.2 mmol/L。
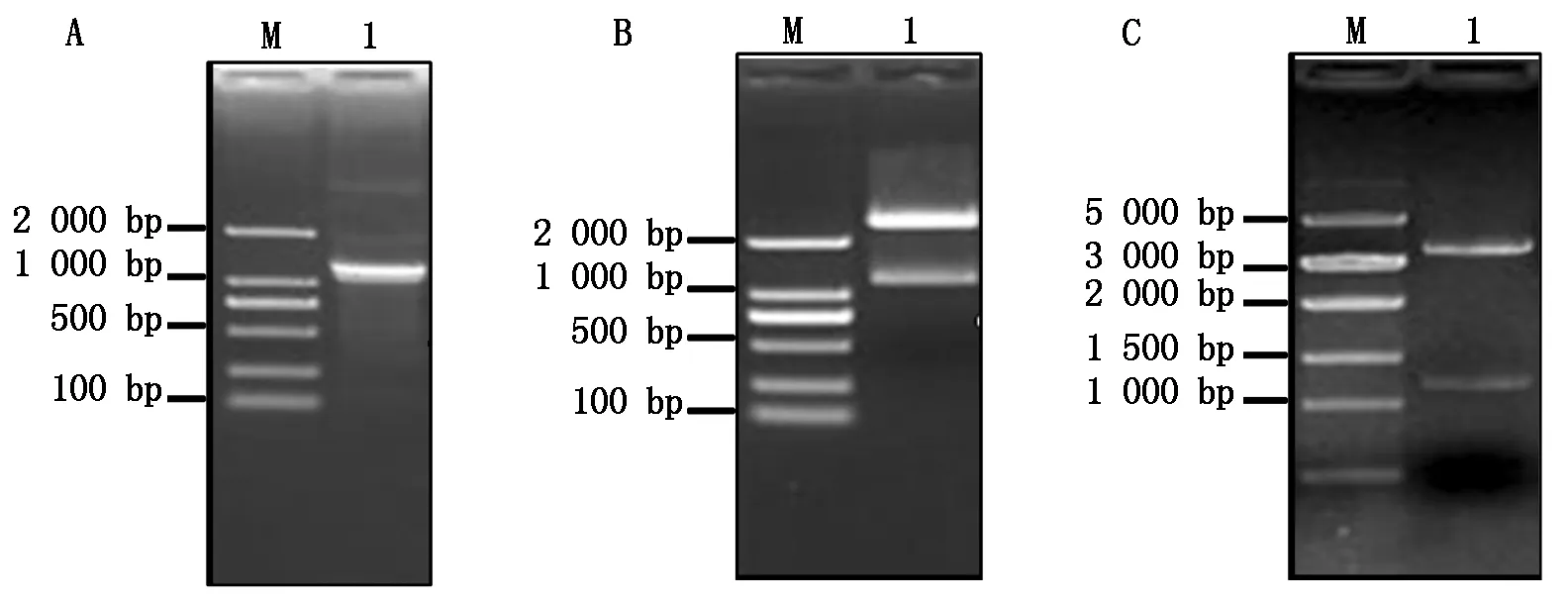
A.BcKMO基因的扩增;B.pMD19-T-BcKMO载体的酶切鉴定;C.pGEX4T-1-BcKMO-GST载体的酶切鉴定。A.PCR amplification of BcKMO;B.Identification of pMD19-T-BcKMO by restriction enzyme digestion;C.Identification of pGEX4T-1-BcKMO-GST by restriction enzyme digestion.
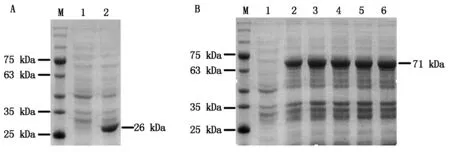
M.蛋白分子量标准;A.诱导表达pGEX4T-1载体;1.IPTG未诱导;2.IPTG诱导GST蛋白;B.pGEX4T-1-BcKMO-GST的诱导表达;1.IPTG未诱导;2~6.IPTG诱导浓度分别为0.1,0.2,0.4,0.8,1.0 mmol/L。M.Protein Marker;A.Expression of pGEX4T-1 with IPTG induction;1.Without IPTG;2.IPTG induces GST proteins;B.Expression of pGEX4T-1-BcKMO-GST with IPTG induction;1.Without IPTG;2-6.Expression of pGEX4T-1-BcKMO-GST with 0.1,0.2,0.4,0.8,and 1.0 mmol/L IPTG.
2.3 不同诱导时间对BcKMO蛋白表达的影响
使用0.2 mmol/L IPTG诱导pGEX4T-1-BcKMO-GST表达,诱导时间分别为4,8,12,16,20 h,收集菌体,以未诱导的菌体作为对照,进行SDS-PAGE检测。结果发现,在不同诱导时间下均可以诱导表达71 kDa的蛋白;当诱导时间为0~12 h时,随着诱导时间的延长,目的蛋白的表达水平逐渐增加;当诱导时间为12~20 h时,目的蛋白的表达水平没有明显变化(图3)。由此确定,蛋白诱导表达的最佳时间为12 h。
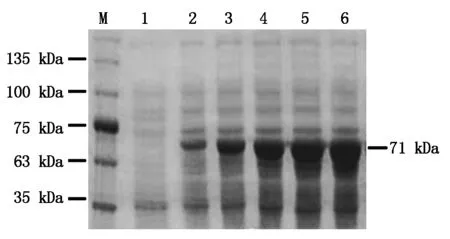
M.蛋白分子量标准;1.IPTG未诱导;2~6.IPTG诱导时间分别为4,8,12,16,20 h。M.Protein Marker;1.Without IPTG;2-6.Expression of BcKMO-GST with IPTG induction for 4,8,12,16,and 20 h.
2.4 BcKMO-GST蛋白的纯化
利用上述得到的最佳诱导条件,进行BcKMO-GST蛋白的诱导表达,并使用ProteinIsoTMGST Resin对收集的上清进行蛋白的纯化。纯化后的蛋白进行SDS-PAGE分析,获得了单一的蛋白条带,大小与目的蛋白大小一致(图4),表明BcKMO-GST蛋白纯化成功。
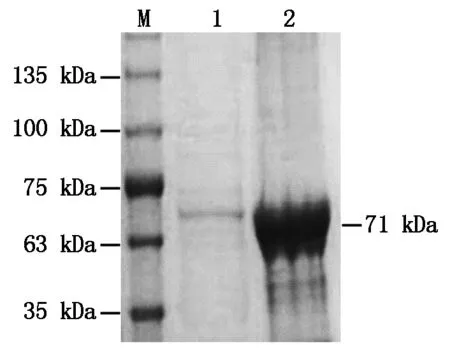
M.蛋白分子量标准;1.纯化的BcKMO-GST蛋白;2.纯化之前的上清液。M.Protein Marker;1.Purified protein BcKMO-GST;2.Supernatant before purification.
2.5 BcKMO-GST蛋白的Western Blot分析
以GST Tag antibody为一抗,goat anti-mouse HRP antibody为二抗,对纯化获得的蛋白进行Western Blot分析。结果发现,纯化的蛋白获得了单一的阳性条带,大小与目的蛋白大小(71 kDa)一致(图5),表明BcKMO蛋白体外诱导表达成功。
3 讨论
外源基因在大肠杆菌体内能否高效表达受到很多因素的影响和限制,如外源基因自身的理化性质、大肠杆菌菌株的类别、所用表达载体种类、诱导物浓度、诱导温度及诱导时间等[22]。一般诱导物选用IPTG[23],低温有利于增强蛋白的稳定性和正确折叠,容易获得可溶性的蛋白,而较高温度则易形成包涵体[24],不利于目的蛋白的纯化,因此,本试验采用28 ℃长时间诱导,降低合成速度,使该蛋白具有充分的时间进行折叠,使二硫键正确配对,使蛋白达到足够的溶解度。本试验通过设置不同IPTG浓度和不同诱导时间进行试验[25],确定了pGEX4T-1-BcKMO-GST融合蛋白诱导表达的最适浓度和最佳时间。超声破碎后使用GST亲和的beads进行目的蛋白的纯化,经SDS-PAGE检测和Western Blot分析确定BcKMO蛋白体外诱导表达成功。但是,蛋白纯化的条件仍需进一步优化,进而获得更高含量的目的蛋白,为后续进行该单加氧酶的酶活力测定及其互作蛋白的筛选奠定基础。
文献报道,犬尿氨酸单加氧酶的稳态动力学参数通过酶活性的初始速率测量获得。KMO催化NADPH依赖的L-犬尿氨酸羟基化成为3-羟基犬尿氨酸,酶促反应能通过接下来的NADPH在340 nm吸光度的降低来监测。速率分析实施采用1 cm路径长度石英微量吸收测定人KMO是37 ℃,酿酒酵母KMO是30 ℃[11]。GST pull-down试验可用于捕获与靶蛋白相互作用的目的蛋白,从而证实2种蛋白之间的相互作用或筛选相应的蛋白。试验后期拟利用Pull-down技术筛选BcKMO蛋白的互作蛋白,为阐明BcKMO及其所在的犬尿氨酸途径与病菌致病力之间的关系提供理论依据。
