CdCO3电子结构与光学属性的第一性原理研究
2017-12-05张水利邵婷婷杨延宁张富春
盛 虹,张水利,邵婷婷,杨延宁,张富春
CdCO3电子结构与光学属性的第一性原理研究
盛 虹1,张水利2,邵婷婷2,杨延宁2,张富春2
(1. 渭南师范学院 数理学院,陕西 渭南 714000;2. 延安大学 物理与电子信息学院,陕西 延安 716000)
采用基于密度泛函理论框架的第一性原理计算方法,利用广义梯度近似方法研究了CdCO3的晶体结构、电子结构和光学属性,理论计算结果表明,CdCO3属于间接宽带隙半导体材料,带隙宽度为2.59 eV,带隙主要由价带顶的Cd 4p、O 2p和导带底的Cd 4p5s轨道能级决定的。而电荷密度结果显示CdCO3晶体是一种离子性较强而共价性较弱的混合键半导体,具有强烈的p轨道与d轨道杂化分布特征。利用精确计算的能带结构和态密度分析了带间跃迁占主导地位的CdCO3材料的光学属性,光学性质的计算结果显示在0~15 eV的能量范围内出现了三个明显的介电峰,吸收带边对应于紫外波段。以上结果对于探索基于CdCO3纳米材料的潜在应用具有重要的理论指导意义,也为精确监测和控制CdCO3材料的生长提供了理论依据。
碳酸镉;第一性原理;电子结构;电荷密度;光学带隙;光吸收
近年来,形貌可控的碳酸盐纳米结构材料CdXO3(X = C, Si, Ge, Sn, Pb)由于其特殊的形态和结构而具有新奇的光学、化学和物理特性,引起人们的广泛研究热情[1]。特别是碳酸镉纳米材料在室温下具有稳定的结构,当材料加热到近500℃时将发生明显的热分解反应,释放出CO2,转变成为CdO材料,从而导致光电属性(电导率、折射率、反射率)发生突变,广泛应用于涤纶材料的中间体和绝缘材料、玻璃助熔剂、催化剂、增塑剂和稳定剂、聚合物电解质以及制备各类镉盐的前驱体材料[2-4]。至今,人们采用水热法、化学浴沉积法(Chemical Bath Deposition, CBD)、超声化学法合成了纳米薄膜、纳米粉体、纳米线、纳米带等不同形貌的CdCO3纳米材料[5]。王卫红等[6]利用镍镉电池产生的废泡沫式镉极板,采用“溶解-萃取-沉淀”方法成功制备出了CdCO3材料;游富英等[7]采用差热分析(Differential Thermal Analysis, DTA)方法系统研究了CdCO3非等温动力学的分解过程,给出了分解反应的表观活化能和反应级数;Khayati等[8]利用热分解法获得CdCO3前驱体,研究制备CdO纳米颗粒;Morenoc等[9]采用化学浴沉积法在玻璃衬底上通过CdS材料成功制备出了CdCO3薄膜材料,通过进一步的退火处理(温度120 ℃),CdCO3材料分解为CdO,并揭示了其光学吸收谱的变化规律。Archer[10]系统研究了CdCO3材料在–268.65~76.85 ℃范围内的热力学属性;Pérez等[11]采用化学浴方法制备出了Pb掺杂CdCO3多晶材料,研究结果显示在1400,850,718 cm–1处出现了明显的红外峰,在670 cm–1处出现了吸收带边;Portillo等[12]也采用CBD方法研究了CdS到CdCO3薄膜材料的转变过程以及它们的光学、结构和形貌特性。
目前,尽管对CdCO3材料的电子结构、热力学属性等方面进行了一些实验研究,但是确切的电学性能、光学性能和掺杂机理仍有许多问题值得去研究,尤其是基于CdCO3材料的发光机理未见详细的报道。因此有必要从理论上对CdCO3材料电子结构和光学属性的内在本质进行研究。本文基于密度泛函理论的第一性原理计算方法系统研究了CdCO3材料的电子结构和光学属性,为金属碳酸盐材料改性研究提供理论依据。
1 理论模型和计算方法
1.1 理论模型
CdCO3室温下是六方晶系,空间群是R-3c,晶格常数为==0.4923 nm,=1.628 nm,90°,=120°。Cd离子占据Wyckoff 2b (0, 0, 0)位置,O离子占据八面体6f (, 0,1/4)位置,C离子占据Wyckoff 2a (0, 0, 1/4)位置,本文构建的CdCO3晶体模型如图1所示。
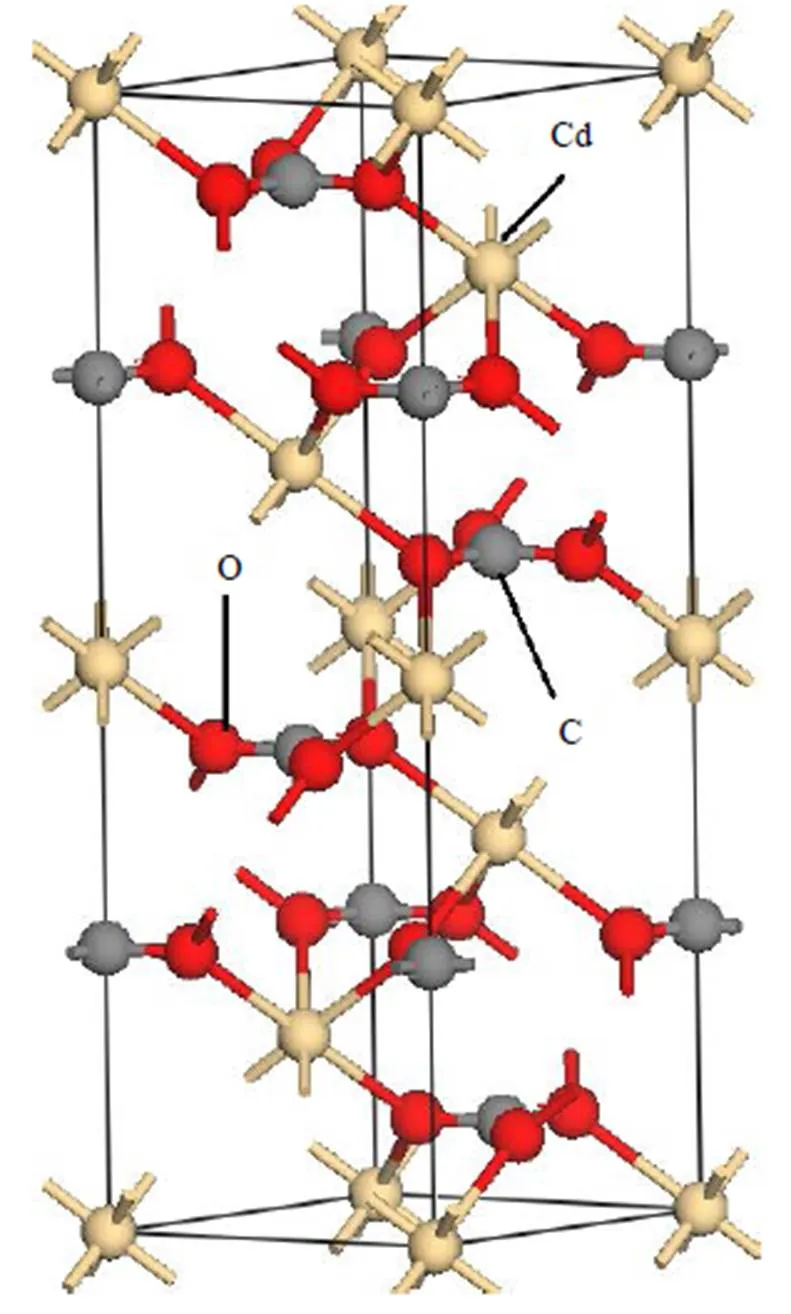
图1 CdCO3模型
1.2 计算方法
本文采用密度泛函理论框架下的第一性原理平面波超软赝势方法,使用VASP[13]软件包进行所有的计算。用广义梯度近似(Generalized Gradient Approximation, GGA)和(Perdew Burke Ernzer, PBE)[14]泛函来处理Cd、C、O原子之间的电子交换关联能。计算过程中,原子的外层价电子选取的组态分别是Cd 4d105s2,C 2s22p2,O 2s22p4,其余均作为芯电子处理。电子波函数用平面波基矢来展开,截断能量为460 eV,布里渊积分设置为10×10×10的Monkhorst-Pack特殊K点,能带结构计算中,布里渊区的积分路径保持和原胞一致,自洽迭代收敛精度不小于1×10–6eV/atom,所有计算都在倒易空间中进行。
1.3 光学性质的理论描述
在光学函数线性响应范围内,固体的宏观光学线性响应函数由光学的复介电函数()=1()+i2()中的复介电函数虚部2()或复折射率()=()+i()决定,其中,和分别为反射系数和消光系数。且:
1=2–2(1)
2= 2(2)
根据Kramer-Kroning[15-16]色散关系和线性响应函数中光学跃迁几率可推导出如下的晶体介电函数虚部2()、实部1()、吸收系数等主要光学函数,具体结果如公式(3)、(4)、(5)和(6)所示:

(4)


式中:、分别表示导带和价带;BZ表示第一布里渊区空间;为倒格矢常数;|e·cv()|2表示光学动量跃迁矩阵元;是角频率参数。C()、V()分别表示导带和价带的轨道能级,这些公式是晶体能带结构和光学性质的理论分析依据,反映轨道能级之间光学跃迁机理。
2 结果与分析
2.1 几何结构优化结果
理论计算的CdCO3材料的晶格参数如表1所示,从表1可以看出,理论计算结果与已有实验值一致[17],晶格参数和误差小于2.2%,体积误差小于5.67%。以上理论计算结果说明本文的计算方法合理,计算结果正确。
表1 CdCO3的几何优化结果

Tab.1 The results of geometry optimization for CdCO3
2.2 电子结构
为了进一步研究CdCO3材料的电子结构和属性,首先利用优化后的晶格参数计算了理想的CdCO3的电子结构,包括能带结构、总体态密度(Total Density of States, TDOS)和分波态密度(Partial Density of States, PDOS),理论计算结果如图2和图3所示。
从图2中可以看出,CdCO3材料是一种间接禁带半导体材料,导带底位于布里渊区的Γ点处,价带顶位于布里渊区中的M点处,理论计算的禁带宽度g是2.59 eV,小于实验值3.87 eV[9],论文虽然采用了广义梯度近似和平面波方法,但此次计算的禁带宽度(g=2.59 eV)仍然偏小,误差是48%。出现上述现象的原因主要是由于局域密度近似(Local Density Approximation,LDA)和GGA都存在着禁带宽度理论计算结果普遍偏低的问题[18],对CdCO3晶体主要是计算中过高地估计了金属镉的4d态能量,导致4d态与O 2p态之间的相互作用增大,价带宽度展宽,禁带宽度变小。但计算结果并不影响对CdCO3晶体电子结构和属性的理论分析,特别是对于导带底Γ点处和价带顶M点处的能带结构的分析。结合CdCO3的总体态密度和分波态密度图可知,CdCO3的价带基本上可以分为两个区域,即0 ~ –3.3 eV的上价带,–4.3~–8.7 eV区的上价带区。CdCO3的上价带能级主要是由Cd 4p和O 2p态贡献的;下价带能级主要是Cd 4d4p部分O 2p和C 2p态共同杂化贡献的;在–19.3 eV处的价带能级主要由Cd 5s和O 2s态贡献的,但由于此处态密度处于深能级,对CdCO3的电子结构和属性影响很小,在文中不做进一步讨论。对于CdCO3晶体的导带能级部分,从图中可以明显看出,导带底态密度主要来源于Cd 4p5s和部分C2p、O 2p能级。因此,CdCO3晶体的禁带宽度主要由价带顶的Cd 4p,O 2p能级和导带底的Cd 4p5s轨道能级决定。

图2 计算的CdCO3能带结构
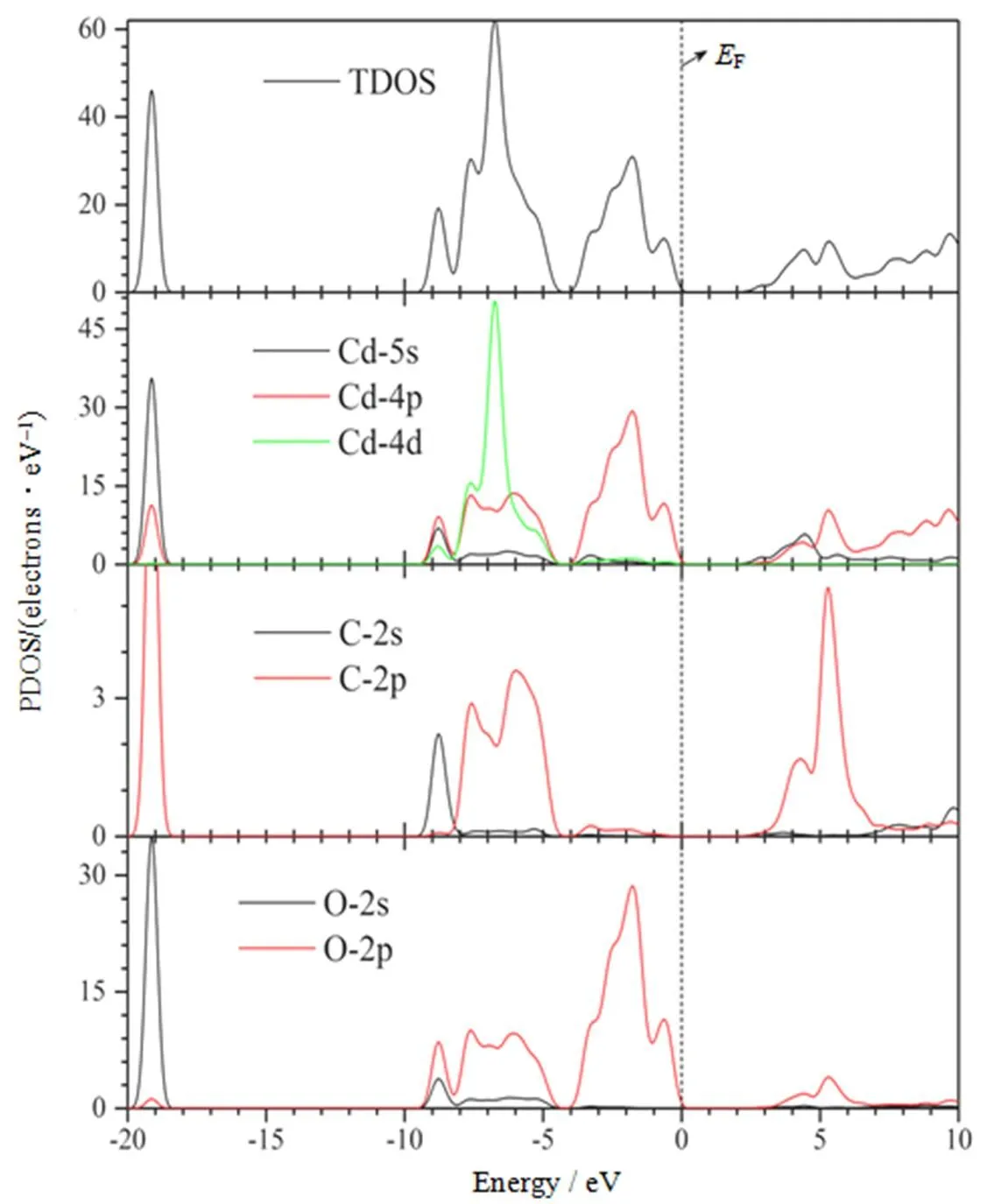
图3 计算的CdCO3态密度
2.3 电荷密度与布局分析
为了进一步分析CdCO3晶体原子之间的成键特性和相互作用特征,本文计算了CdCO3晶体的差分电荷密度、布局和电荷转移性能,理论计算的差分电荷密度如图4所示,电荷转移特性和布局如表2所示。从图4和表2的布局可以看出,CdCO3晶体中Cd离子与O离子是一种混合价化合物半导体材料,主要以离子键为主,同时兼有共价键成分,C离子和O离子主要以共价键为主,而O离子与O离子主要以离子键为主。C原子周围的电荷密度出现了明显的sp3杂化效应,O原子周围出现了具有P轨道的电子分布特征,Cd原子周围形成了4d轨道极化电子分布特征,具有强烈的C和O 2p轨道与Cd 4d轨道能级杂化分布特征。整体差分电荷密度具有较强的离散性特征,从图4可以清楚地看到电子发生了重新分配和移动,每个O原子失去了0.66个电子,Cd原子和C原子分别得到了1.25个和0.71个电子。以上理论结果表明,CdCO3晶体是一个离子性较强而共价性较弱的混合键宽禁带半导体材料,这与上面计算的能带结构和态密度计算结果一致。理论计算结果对于揭示CdCO3晶体材料的几何结构和属性具有重要的理论意义。
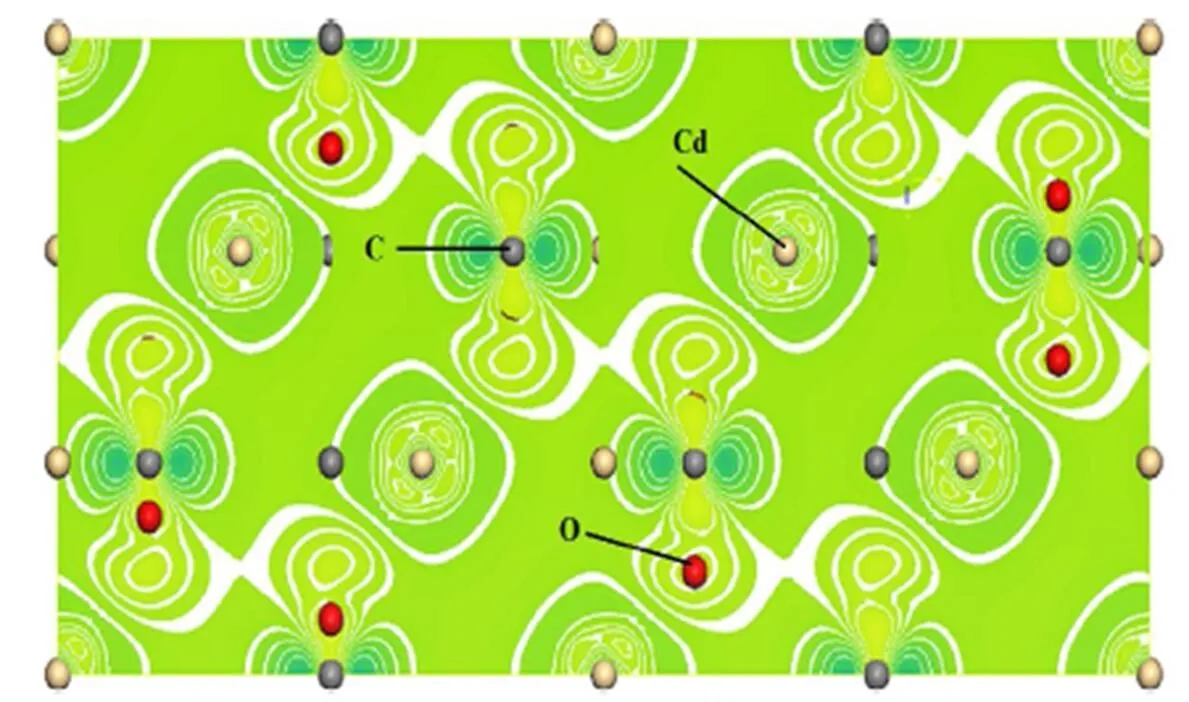
图4 计算的CdCO3差分电荷密度
表2 计算的CdCO3电荷和布局分布
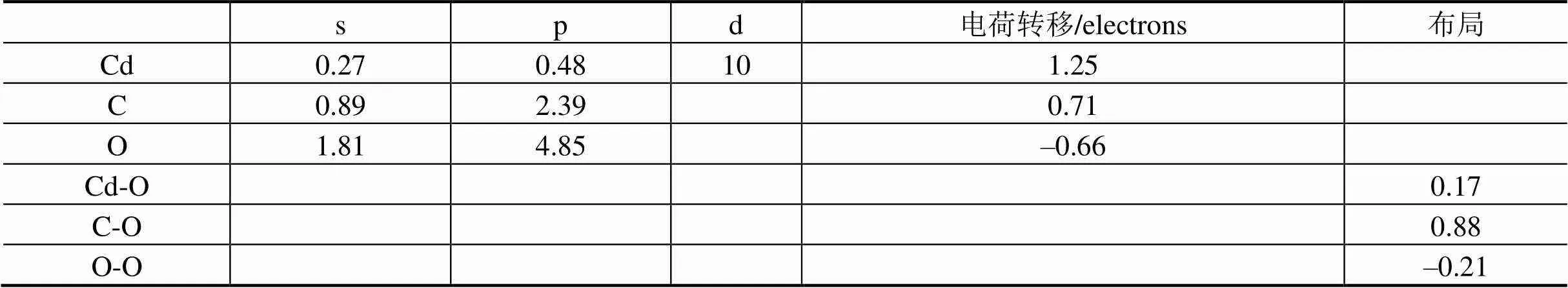
Tab.2 Calculated charge distribution and population of CdCO3
2.4 光学性质
CdCO3是一类间接宽带隙金属碳酸盐半导体材料,其光谱属性决定于轨道能级之间的电子跃迁。图5是计算得到的CdCO3复介电函数随光子能量变化的能谱图。从图中可以看出,理论计算得到的静态介电常数0=2.68,在低能段(0~5.5 eV),介电函数实部随能量的增加而缓慢增大,当能量大约为5.5 eV时达到最大值。在高能段(大于5.5 eV),介电函数实部随光子能量增大而急剧减小,即CdCO3在高能段(大于5.5 eV)带间电子跃迁光吸收显著增强,在9.0 eV和12.5 eV出现了两个明显的吸收跃迁峰。另外,从图中可以看出,CdCO3材料介电函数虚部出现了3个比较明显的介电峰,分别位于6.3,9.5和13.3 eV。特别是CdCO3晶体的价带顶和导带底附近具有很多折叠和弥散的轨道能级,在布里渊区K空间内,CdCO3晶体轨道能级除去选择定则规定的禁戒跃迁外,其他轨道能级都可以发生价带到导带的电子跃迁。结合理论计算的分波态密度和能带结构图,在2.59 eV处开始出现光吸收,对应于价带顶到导带底的间接跃迁阈,它主要来源于O 2p和Cd4p态到Cd 5s态跃迁;在6.3 eV附近出现了一个陡峭的介电峰,它主要来源于上价带Cd 4p和O 2p在3.2 eV态密度峰到未占据导带之间的跃迁;在13.3 eV附近也出现了一个较强的介电吸收峰,它主要来源于下价带Cd 4d态到导带的Cd 5s 和O 2p态的跃迁。以上理论分析结果与目前得到的一些实验结果是吻合的[9,12],对揭示CdCO3材料的光学性质的内在属性具有重要的理论指导意义。
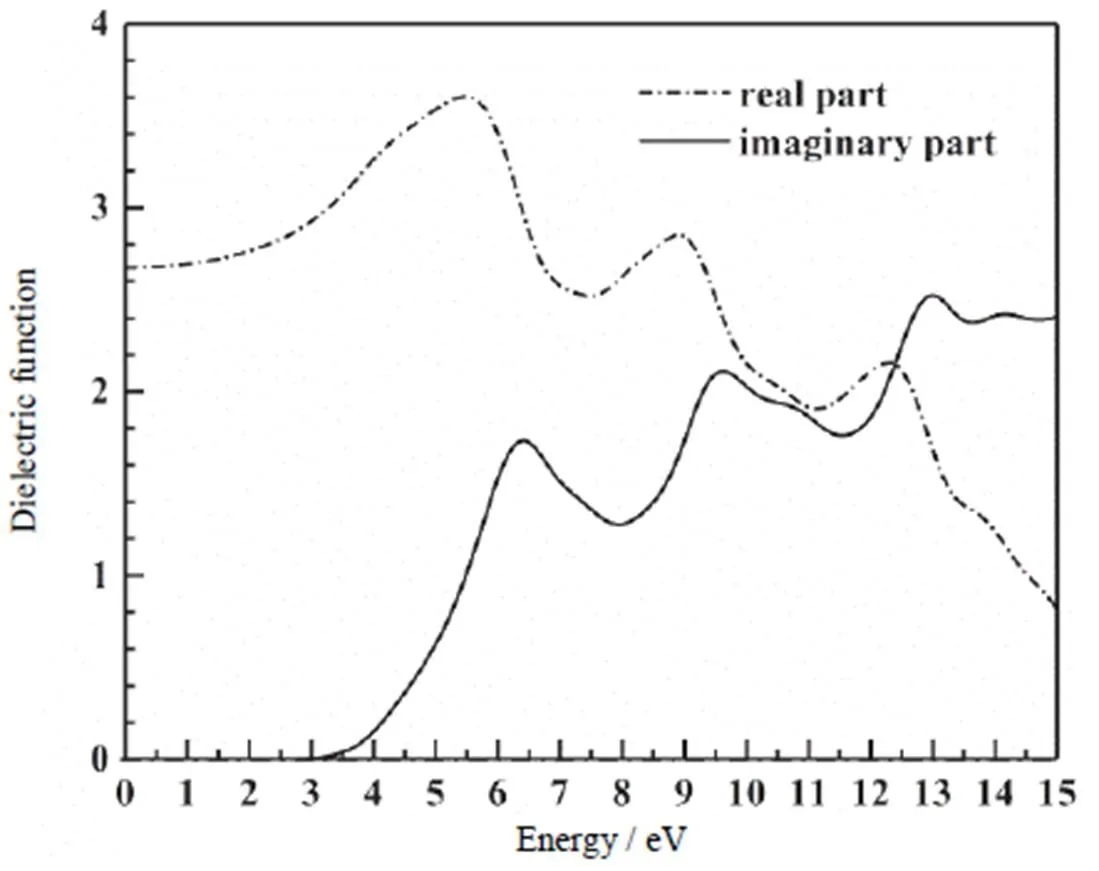
图5 CdCO3的介电函数
3 结论
采用基于密度泛函理论的第一性原理方法计算了CdCO3的几何结构、能带结构、电荷密度、态密度和光学属性,分析了电子结构与光学性质的内在机理,结果表明:CdCO3理论预测是一种间接宽带隙半导体材料,理论计算的间接带隙为2.59 eV;CdCO3是一种混合价化合物半导体材料,主要以离子键为主,同时兼有共价键成分,CO32–离子团周围的电荷密度出现了明显的sp3杂化效应;通过精确计算的光学介质跃迁矩阵元,分析了CdCO3电子结构与光学属性之间的本质关系,揭示了介电吸收峰跃迁机理,对介电谱图的峰值进行了指认和判别。理论预测CdCO3是一种在光电领域具有潜在用途的纳米功能材料,也为合成CdCO3纳米材料提供了理论依据。
[1] BARBOZA C A, HENRIQUES J M, ALBUQUERQUE E L, et al. (X=C, Si, Ge, Sn, Pb) electronic band structures [J]. Chem Phys Lett, 2009, 480(4/5/6): 273-277.
[2] ANTIPOV A A, SHCHUKIN D, FEDUTIK Y, et al. Carbonate microparticales for hollow polyelectrolyte capsules fabrication [J]. Coll Surf A Physicochem Eng Aspects, 2003, 224: 175-183.
[3] NOROOZIFAR M, MOTLAGH M K, HOSSEINI S N. Flow injection analysis-flame atomic absorption spectrometry system for indirect determination of cyanide using cadmium carbonate as a new solid-phase reactor [J]. Anal Chim Acta, 2005, 528(2): 269-273.
[4] GUO Z, LI M, LIU J. Highly porous CdO nanowires: Preparation based on hydroxy- and carbonate-containing cadmium compound precursor nanowires, gas sensing and optical properties [J]. Nanotechnology, 2008, 19(24): 245611.
[5] JIA Z Y, TANG Y W, LUO L J, et al. Shape-controlled synthesis of single-crystalline CdCO3and corresponding porous CdO Nanostructures [J]. Cryst Growth Des, 2008, 8(7): 2116-2120.
[6] 王卫红, 陈志传, 李玉清, 等. 镍镉电池废镉极板制备碳酸镉的研究[J]. 环境工程, 2001, 19(2): 33-36.
[7] 游富英, 刘妍. 碳酸镉非等温动力学参数的确定[J]. 邯郸师专学报, 2004, 14(3): 62-63.
[8] KHAYATI G R, SHAHCHERAGHI S H, LOTFI V, et al. Reaction pathway and kinetics of CdO nanoparticles prepared from CdCO3precursor using thermal decomposition method [J]. Trans Nonferrous Met Soc Chin, 2016, 26(4): 1138-1145.
[9] MORENO1 O P, ROJAS1 A V, TECORRALCO1 J H, et al. Transformation on CdS→CdCO3thin films by chemical bath and CdCO3→CdO annealing thermal in atmosphere Air [J]. J Mater Sci Eng, 2011, A1: 991-999.
[10] ARCHER D G. Thermodynamic properties of synthetic otavite, CdCO3(Cr): enthalpy increment measurements from 4.5 K to 350 K [J]. J Chem Eng Data, 1996, 41: 852-858.
[11] PÉREZ R G, MORENO O P, PORTILLO M C, et al. Synthesis of CdCO3in situ-doped-Pb2+grown by chemical bath [J]. Mater Lett, 2015, 160: 488-490.
[12] PORTILLO M C, SANCHEZA A S G, DIAZ G J, et al. Optical, structural and morphological properties of CdS-CdCO3films [J]. Rev Mex Fis, 2015, 61: 83-87.
[13] KRESSE G, FURTHMÜLLER J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set [J]. Phys Rev B, 1996, 54: 11169.
[14] PERDEW J P, BURKE K, ERNZERHOF M. Generalizd gradient approximation made simple [J]. Phys Rev Lett, 1996, 77(18): 3865-3868.
[15] 谢希德, 陆栋. 固体能带理论 [M]. 上海: 复旦大学出版社, 1998.
[16] 沈学础. 半导体光谱和光学性质(第二版) [M]. 北京: 科学出版社, 1992.
[17] BORODIN V L, LYUTIN V I, ILYUKHIN V V, et al. Isomorphous calcite-otavite series [J]. Dokl Akad Nauk SSSR, 1979, 245: 1099-1101.
[18] CHING W Y, XU Y N, WONG K W. Ground-state and optical properties of Cu2O and CuO crystals [J].Phys Rev B, 1998,40(11): 7684-7695.
(编辑:陈丰)
First-principles study on electronic structure and optical properties of CdCO3
SHENG Hong1, ZHANG Shuili2, SHAO Tingting2, YANG Yanning2, ZHANG Fuchun2
(1. College of Mathematics & Physics, Weinan Normal University, Weinan 714000, Shaanxi Province, China; 2. College of Physics and Electronic Information, Yan’an University, Yan’an 716000, Shaanxi Province, China)
The crystal structure, electronic structure and optical properties of CdCO3were studied by using the first-principles based on the density functional theory (DFT) within the generalized gradient approximation (GGA). The theoretical results indicate that the CdCO3is an indirect bandgap semiconductor material and the band gap is 2.59 eV, which is mainly determined by Cd 4p, O 2p orbital energy levels at the top of valence band and Cd 4p, 5s orbital energy levels at the bottom of conduction band. The results of charge density show that CdCO3crystal is a hybrid bond semiconductor strong in ionicity bond and weak in covalent, characterized by intense p-d hybrid orbitals. The optical properties of the CdCO3in which the band-to-band transition are at leading status are analyzed by the precisely calculated band structure and density of state (DOS). The results of the optical properties show that obvious three dielectric peaks appear in energy range from 0 eV to 15 eV, and the absorb edges are located in the ultraviolet region. The above results have important theoretical significance for us to explore the potential applications based on CdCO3material and offer theoretical reference to precisely monitor and control the growth of CdCO3material.
CdCO3; the first-principles; electronic structure; charge density; optical band gap; optical absorption
10.14106/j.cnki.1001-2028.2017.12.009
O643.1
A
1001-2028(2017)12-0042-05
2017-11-03
张富春
国家自然科学基金资助(No. 61664008); 陕西省高水平大学建设项目赞助(No. 2015SXTS02);渭南师范学院资助项目(No. 15YKS006, No. 2015JYKX018)
张富春(1972-),男,陕西定边人,教授,主要从事半导体材料与器件的研究,E-mail: zhangfuchun72@163.com;
盛虹(1971-),女,陕西渭南人,副教授,主要从事物理教学理论及物理实验研究,E-mail: wnshenghong@163.com 。
2017-11-30 14:11
网络出版地址: http://kns.cnki.net/kcms/detail/51.1241.TN.20171130.1411.008.html
