多脏器型假性醛固酮减少症Ⅰ型1例并文献复习
2017-12-02邹亮燕曾丽春蒋思远琳周文浩陈丽萍王来栓
邹亮燕 曾丽春 蒋思远 陈 乡 杨 琳周文浩陈丽萍 王来栓,
多脏器型假性醛固酮减少症Ⅰ型1例并文献复习
邹亮燕1,4曾丽春2,4蒋思远1陈 乡3杨 琳3周文浩3陈丽萍2王来栓1,3
目的 报告1例SCNN1A基因杂合突变所致的多脏器型假性醛固酮减少症Ⅰ型(PHAⅠ),为PHAⅠ患儿的早期诊断、治疗等提供依据。方法 总结患儿的临床表型、实验室检查、基因测序结果和诊治经过,并对多脏器型PHAⅠ行文献复习。结果 患儿男,17 d,因“发现电解质紊乱6 d”入院,主要表现为低钠血症、高钾血症和代谢性酸中毒。经补钠、氢化可的松和胰岛素等治疗效果不理想。核心家系全外显子检测发现SCNN1A复合杂合变异c.1439+1Ggt;C和c.104delC(p.P35LfsTer14)。c.1439+1Ggt;C源于患儿父亲,是人类基因突变数据库(HGMD)已报道的多脏器型PHAⅠ型的致病突变;c.104delC(p.P35LfsTer14)源于患儿母亲,系新发现的突变。明确诊断多脏器型PHAⅠ后拟予口服降钾树脂治疗,患儿家属放弃治疗,出院后第4 d患儿死亡。检索Pubmed、中国期刊全文数据库、万方数据库、中文科技期刊数据库和中国生物医学文献数据库,检索时间从建库至2017年9月4日,14篇文献(13篇英文,1篇中文)共报告33例多脏器型PHAⅠ型患儿,与本文1例合并后共34例。34例均有肾脏表现。除3例SCNN1A纯合突变及1例SCNN1G杂合突变在病程及随访中未提及肾外表现外,余病例均报道累及肾外其他系统,其中脱水样改变和累及呼吸道最常见,发育迟缓和消化道次之。4例死于高血钾所致心脏骤停,均为SCNN1A突变,除本文报告的1例为杂合突变外均为纯合突变。 结论 顽固性高钾血症、低钠血症和代谢性酸中毒需考虑PHA,基因测序可协助诊断。
SCNN1A基因; 醛固酮减少症; 高钾血症; 低钠血症; 代谢性酸中毒; 基因诊断
1 病例资料
患儿男,17 d。因“发现电解质紊乱6 d”于2017年7月21日到复旦大学附属儿科医院(我院)就诊。
患儿系G3P3,胎龄37周,2017年7月4日于当地医院顺产娩出,否认抢救窒息史,出生体重3 000 g,1和5 min Apgar评分均为10分。生后以“高危儿”入当地医院,因低钾、低钠治疗效果不佳入我院就诊。病程中患儿无发热、抽搐,无青紫、呼吸困难,无呕吐、腹胀,纳奶一般,大小便未见明显异常。患儿已注射维生素K1,接种乙肝疫苗、卡介苗,新生儿筛查未见异常,听力筛查不详。
患儿父母体健,否认近亲结婚,否认遗传病史,母孕产史3-0-0-1,分别于2012和2014年产两女孩,均在生后10 d因“高钾血症”入当地医院治疗,1个月内均死亡。
查体:肛温36.5 ℃,脉搏138·min-1,呼吸42·min-1,血压51/33 mmHg。头围34.0 cm,胸围32.5 cm,腹围33.0 cm,身长50.0 cm,体重2 565 g。神志清,反应欠佳,哭声尚可。全身皮肤苍白,表浅淋巴结未触及。头颅无畸形,面容无特殊,前囟平软,1.5 cm×1.5 cm。心音有力,心前区未闻及杂音。双肺呼吸音粗,未及干湿啰音。肝、脾未触及肿大,腹平软,肠鸣音3·min-1。阴茎无畸形,阴囊颜色偏深,阴囊内可触及黄豆大小睾丸组织,原始反射可引出。
实验室检查:血气K+8.3 mmol·L-1,Na+123.0 mmol·L-1,BE -10.8 mmol·L-1,HCO3-18.0 mmol·L-1,PCO219.0 mmHg,pH 7.45。血、尿、粪常规未见异常。血串联质谱阴性,尿串联质谱2-酮戊二酸高。皮质醇、促肾上腺皮质激素(ACTH)、17α-羟孕酮、睾酮、孕酮、脱氢表雄酮、肾素、游离甲状腺素(FT4)和促甲状腺激素(TSH)在正常范围,血管紧张素Ⅱ(550 pg·mL-1)升高,醛固酮(1 254.72 μg·mL-1)升高。
影像学检查:B超显示双肾、肾上腺、输尿管和膀胱无明显异常;肾上腺增强CT未见明显异常;心电图显示窦性心动过速、T波改变(V1直立)。
诊治经过:图1为诊断、治疗和随访等重要临床信息时间轴。入院时考虑先天性肾上腺皮质增生症(CAH),予氢化可的松治疗效果不理想,醛固酮明显增高,ACTH、皮质醇等在正常值范围,影像学无阳性证据。鉴于患儿两姐姐均因“高血钾症”死亡,建议行基因检测,患儿存在SCNN1A基因的复合杂合变异。诊断为多脏器型PHAⅠ型。停氢化可的松,予10%NaCl 3.5 mL,q3 h 口服,5%NaHCO3和胰岛素降血钾,同时予低钾奶粉喂养,血钾控制仍不稳定。建议予口服降钾树脂治疗,但患儿家属要求出院。 出院后第4天,患儿因高钾合并肺部感染死亡。
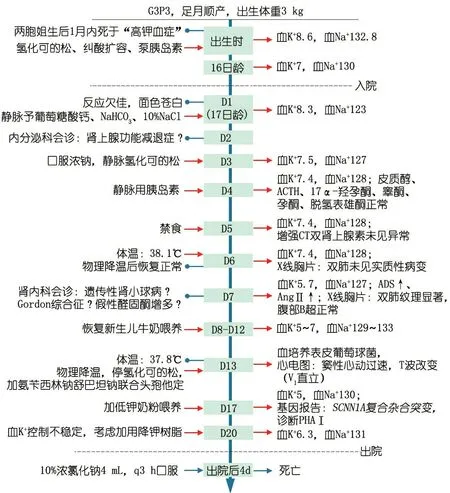
图1本文病例重要临床信息时间轴
注 血K+、Na+单位:mmol·L-1,ADS:醛固酮,AngⅡ:血管紧张素Ⅱ
取得患儿父母知情同意后,采集患儿及其父母外周静脉血2 mL,抽提基因组DNA(德国Qiagen公司mini blood全血试剂盒),捕获外显子、建库[安捷伦科技(中国)有限公司SureSelctHuman ALL Exon试剂盒],对全基因组编码区外显子进行测序(美国Illumina公司HiSeq2000)。通过数据质量控制、变异频率和变异类别的筛选以及与疾病的关系,锁定可能的致病突变。
图2显示,患儿存在SCNN1A基因的复合杂合变异c.1439+1Ggt;C和c.104delC(p.P35LfsTer14)。c.1439+1Ggt;C来自患儿父亲,是人类基因突变数据库(HGMD)已报道的多脏器型PHAⅠ型的致病突变[1]。该突变发生于剪切区,造成SCNN1A基因9号内含子的保留。c.104delC(p.P35LfsTer14)来自患儿母亲,未在HGMD报道,也未在千人基因组计划、ExAC数据集及我院分子诊断中心内部数据库中检索到,为罕见变异。该变异为移码变异,考虑为有害变异。
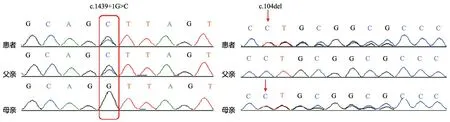
图2患儿及其父母SCNN1A基因检测
2 文献复习
从Pubmed、中国期刊全文数据库、万方数据库、中文科技期刊数据库和中国生物医学文献数据库中检索已报道的PHA Ⅰ病例,检索时间从建库至2017年9月4日。Pubmed检索式为“(systemic pseudohypoaldosteronism type Ⅰ[title/abstract/key word])”;以CBM为例,中文检索式为“假性醛固酮减少症Ⅰ型 OR Cheek-Perry综合征”。采用主题和自由途径结合方式检索,筛选出确诊的多脏器型PHAⅠ病例的文献,排除重复报道病例。
共检索到13篇英文文献[2-14]和1篇中文文献[15],报告33例多脏器型PHAⅠ型患儿,与本文1例合并后共34例患儿的平均发病年龄7.6日龄,平均随访年龄6.1岁。34例病程中都有不同程度高钾低钠的电解质紊乱,主诉以“脱水”和“呕吐”最多,有以“大疱性皮炎”、“胃纳差”、“体重不增”、“生长受限”和“反应差”等就诊者。1例未发现基因突变,其余33例中,26例为SCNN1A突变(20例纯合突变,6例杂合突变),5例为SCNNIB突变(均为纯合突变),2例SCNN1G突变(1例纯合突变,1例杂合突变)。34例均有肾脏表现, 除3例SCNN1A纯合突变及1例SCNN1G杂合突变在病程及随访中未提及肾外表现外,余病例均累及不同肾外系统。7例(5例SCNNIA突变,1例SCNNIB突变,1例未检测到突变)检测了汗液或汗液和唾液氯化物,均升高。4例死于高血钾所致心脏骤停,均为SCNN1A突变,3例为纯合突变,1例为杂合突变(本文)。不同基因型临床表型及预后比较见表1。
3 讨论
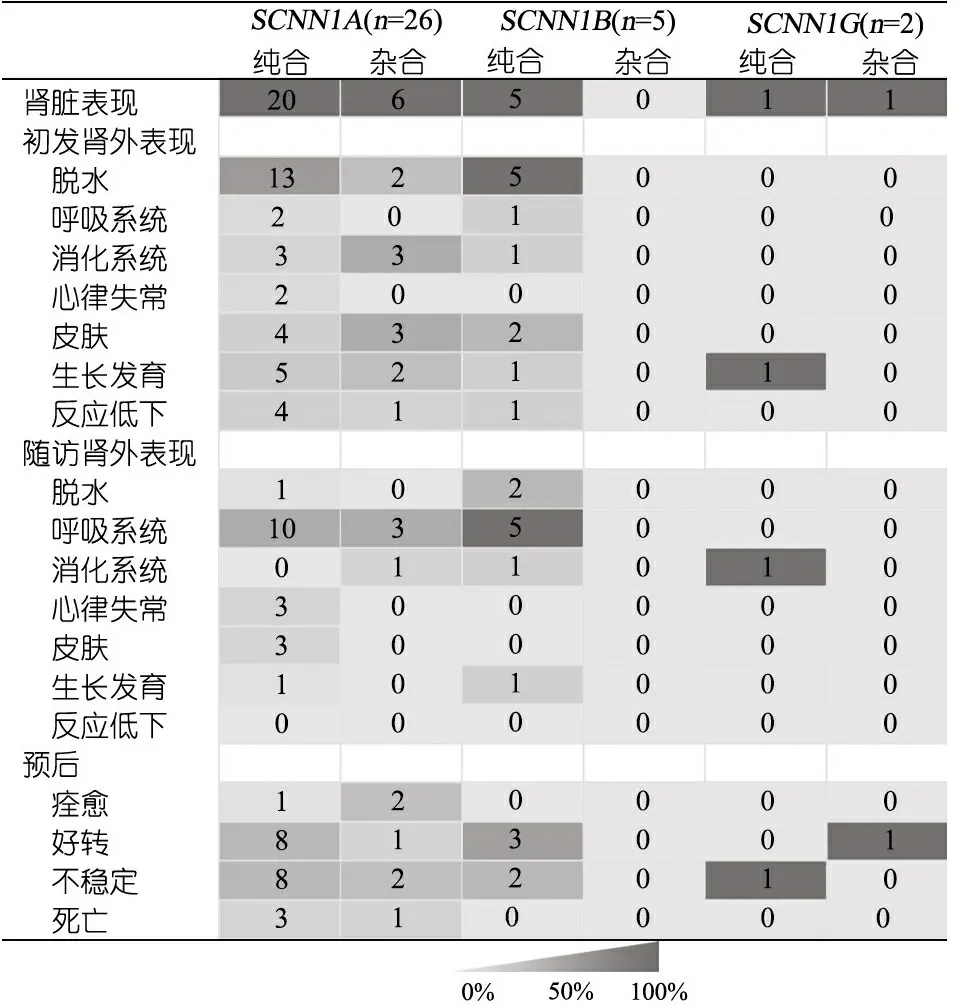
表1 多脏器型假性醛固酮减少症Ⅰ型不同基因型患儿肾外表现和预后
1991年,Hanukoglu通过家系调查发现PHAⅠ可分为多脏器型和肾型[16]。多脏器型PHAⅠ系常染色体隐性遗传,其发病机制与编码上皮钠离子通道(ENaC)亚基的基因突变有关。ENaC是由α、β和γ亚基组成的多聚体,可重吸收Na+,对维持Na+平衡、调节细胞外液容量和血压很重要。α、β、γ亚基分别由SCNN1A(12p13.31)、SCNN1B(16p12.1)和SCNN1G(16p12.1)编码[17]。ENaC主要分布于肾脏远曲小管、呼吸道、外分泌腺、结肠远端和皮肤等的上皮组织[18]。多脏器型PHAⅠ表现为脱水、低钠血症、高钾血症和代谢性酸中毒等电解质紊乱,也可合并呼吸道感染、胆汁淤积和皮疹等[19]。肾型PHAⅠ系常染色体显性遗传,与NR3C2[4q31.1,编码盐皮质激素受体(MCR)]突变有关[20],导致肾小管MCR对醛固酮不敏感,丢失钠盐,病变局限于肾脏,表现为脱水、低钠血症和高钾血症等电解质紊乱,但病情较轻且随年龄增长可自行缓解[21]。
本文患儿因“发现电解质紊乱6 d”入院,表现为低钠血症、高钾血症和代谢性酸中毒等电解质紊乱,入院诊断为“肾上腺皮质增多症”,予氢化可的松治疗无效。基因检测结果示SCNN1A复合杂合变异,包括剪切位点突变c.1439+1Ggt;C和碱基缺失突变c.104delC(p.P35LfsTer14),最终明确诊断为多脏器型PHAⅠ。表1为不同基因型与临床表型及预后的总结与比较,发现报道的34例多脏器型PHAⅠ患儿均存在不同程度的高钾低钠、血肾素及醛固酮升高等肾脏表现。除3例SCNN1A纯合突变及1例SCNN1G杂合突变在病程及随访中未提及肾外表现外,余病例均报道累及肾外不同系统,总结后发现,SCNN1A突变初发肾外表现以脱水样改变最常见(58%),其次为累及皮肤及影响生长发育(各占27%),后期随访中累及呼吸系统的概率最大(50%),而脱水可纠正、生长发育可追赶;SCNN1B突变初发肾外表现均有脱水样改变,其次累及皮肤(40%),后期随访中显示5例呼吸系统全部累及;SCNN1G仅报道2例,肾外表现仅出现于纯合突变类型中。此外,表现为心律失常的5例均为SCNN1A纯合突变,余病例均未报道有心律失常的表现,提示不同突变基因可能导致不同的病变累及范围,具体机制需进一步研究。由此可见,多脏器型PHAⅠ患儿在新生儿期起病,急性期面临的问题主要是电解质紊乱,特别应警惕高血钾,在疾病的早期累及呼吸系统等肾外系统的表现尚不明显,随着年龄的增长,电解质紊乱问题得以控制,远期随访发现,肾外脏器表现可能逐一显现,影响患儿生活质量及增加住院概率。本文患儿以电解质紊乱起病,住院后期出现肺部感染,出院后死于高血钾合并肺部感染,但缺乏汗腺及唾液腺氯化物检测结果。
多脏器型PHAⅠ低钠高钾、代谢性酸中毒的表现易与CAH 相混淆,还应与继发型PHA、肾型PHAⅠ和Ⅳ型肾小管酸中毒相鉴别。CAH系常染色体隐性遗传病,是由肾上腺激素合成过程中所需酶的先天性缺陷所导致的一组疾病,最常见的为21-羟化酶缺乏导致皮质醇分泌不足、失盐和雄激素过多,此外,女性患儿出生时伴假两性畸形,男性患儿生殖器正常或阴茎较大、阴囊颜色深,增强CT可发现肾上腺皮质增生,经过激素治疗可纠正失盐症状[22]。继发型PHA是由尿路感染、泌尿道不畅或畸形、药物引起暂时性醛固酮降低,尿常规异常、尿培养阳性、B超发现尿路畸形、肾盂积水扩张有助于诊断,经抗感染、纠正原发畸形后,症状得以缓解,其发病机制尚未明确,可能的原因是尿路感染或畸形导致引流障碍时,某些细胞因子如转化因子、肿瘤坏死因子、白介素-1和白介素-6下调醛固酮受体[23]。肾型PHAⅠ与多脏器型PHAⅠ较难区分,汗液和唾液中钠离子升高常提示为多脏器型,确诊需基因分析。Ⅳ型肾小管酸中毒多见于某些轻、中度肾功能不全的肾病患者,醛固酮分泌减少或远端肾小管对醛固酮反应减弱可能起重要致病作用,由于远端肾小管泌H+障碍,尿NH4+减少[24]。表2总结了以上5种疾病的实验室检查与影像等检查特征。

表2 多脏器型PHAⅠ与其他疾病的鉴别诊断
注 -:在正常范围
本文文献复习显示,34例PHAⅠ患儿中3例(8.8%)达到临床痊愈,2例为SCNN1A杂合突变,1例为SCNN1A纯合突变; 13例(38.2%)长期补钠及口服降钾树脂,病情保持平稳;14例(41.2%)在长期补钠及口服降钾树脂情况下仍间断出现失盐症状、呼吸道感染或皮疹等症状;4例(11.8%)患儿死于高血钾所致心跳骤停,3例为SCNN1A纯合突变,本文病例为SCNN1A杂合突变。回顾3例SCNN1A纯合突变死亡患儿的诊治方案,与其他患儿相比并无明显特殊,提示多脏器型PHAⅠ患儿的预后可能与本身突变基因的类型和位置有关[6, 25]。Hanukoglu等[6]长期随访4例多脏器型PHAⅠ,除1例未发现突变外,2例为ENaCα、β亚基纯合突变,1例为α亚基杂合突变,4例的尿钠和尿钾水平随年龄增长都恢复正常,但杂合突变者所需的治疗钠量最低,而纯合突变患者病情相对不稳定,住院率和肺部感染率更高。多脏器型PHAⅠ不同突变基因和突变类型与临床表型和预后的关系及机制仍待进一步研究。
[1] Wang J, Yu T, Yin L, et al. Novel mutations in theSCNN1Agene causing Pseudohypoaldosteronism type 1. PloS one, 2013, 8(6): e65676
[2] Schaedel C, Marthinsen L, Kristoffersson AC, et al. Lung symptoms in pseudohypoaldosteronism type 1 are associated with deficiency of the alpha-subunit of the epithelial sodium channel. J Pediatr, 1999, 135(6): 739-745
[3] Thomas CP, Zhou J, Liu KZ, et al. Systemic pseudohypoaldosteronism from deletion of the promoter region of the human Beta epithelial na(+) channel subunit. Am J Respir Cell Mol Biol, 2002, 27(3):314-419
[4] Saxena A, Hanukoglu I, Saxena D, et al. Novel mutations responsible for autosomal recessive multisystem pseudohypoaldosteronism and sequence variants in epithelial sodium channel alpha-, beta-, and gamma-subunit genes. J Clin Endocrinol Metab, 2002, 87(7):3344-3350
[5] Edelheit O, Hanukoglu I, Gizewska M, et al. Novel mutations in epithelial sodium channel (ENaC) subunit genes and phenotypic expression of multisystem pseudohypoaldosteronism. Clin Endocrinol (Oxf), 2005, 62(5):547-553
[6] Hanukoglu A, Edelheit O, Shriki Y, et al. Renin-aldosterone response, urinary Na/K ratio and growth in pseudohypoaldosteronism patients with mutations in epithelial sodium channel (ENaC) subunit genes. J Steroid Biochem Mol Biol, 2008, 111(3-5): 268-274
[7] Belot A, Ranchin B, Fichtner C, et al. Pseudohypoaldosteronisms, report on a 10-patient series. Nephrol Dial Transplant, 2008, 23(5):1636-1641
[8] Schweiger B, Moriarty MW, Cadnapaphornchai MA. Case report: severe neonatal hyperkalemia due to pseudohypoaldosteronism type 1. Curr Opin Pediatr, 2009, 21(2):269-271
[9] Adachi M, Asakura Y, Muroya K, et al. Increased Na reabsorption via the Na-Cl cotransporter in autosomal recessive pseudohypoaldosteronism. Clin Exp Nephrol, 2010, 14(3):228-232
[10] Dirlewanger M, Huser D, Zennaro MC, et al. A homozygous missense mutation inSCNN1Ais responsible for a transient neonatal form of pseudohypoaldosteronism type 1. Am J Physiol Endocrinol Metab, 2011, 301(3):E467-473
[11] Dogan CS, Erdem D, Mesut P, et al. A novel splice site mutation of the beta subunit gene of epithelial sodium channel (ENaC) in one Turkish patient with a systemic form of pseudohypoaldosteronism Type 1. J Pediatr Endocrinol Metab 2012, 25(9-10):1035-1039
[12] Ekinci Z, Aytac MB, Cheong HI. A case ofSCNN1Asplicing mutation presenting as mild systemic pseudohypoaldosteronism type 1. J Pediatr Endocrinol Metab, 2013, 26(11-12): 1197-1200
[13] Welzel M, Akin L, Buscher A, et al. Five novel mutations in theSCNN1Agene causing autosomal recessive pseudohypoaldosteronism type 1. Eur J Endocrinol, 2013, 168(5): 707-715
[14] Nur N, Lang C, Hodax JK, et al. Systemic Pseudohypoaldosteronism Type I: A Case Report and Review of the Literature. Case Rep Pediatr, 2017, 2017:7939854
[15] 沈烨, 沈永年, 王剑, 等. 假性醛固酮减少症Ⅰ型患儿2例临床与基因分析. 临床儿科杂志, 2011, 29(10): 983-987
[16] Hanukoglu A. Type I Pseudohypoaldosteronism Includes Two Clinically and Genetically Distinct Entities with either Renal orMultiple Target Organ Defects. J Clin Endocrinol Metab,1991, 73(5): 936-944
[17] Meisler MH, Barrow LL, Canessa CM, et al. SCNN1, an epithelial cell sodium channel gene in the conserved linkage group on mouse chromosome 6 and human chromosome 12. Genomics, 1994, 24(1):185-186
[18] Duc C, Farman N, Canessa CM, et al. Cell-specific expression of epithelial sodium channel alpha, beta, and gamma subunits in aldosterone-responsive epithelia from the rat: localization by in situ hybridization and immunocytochemistry. J Cell Biol, 1994, 127(6):1907-1921
[19] Eliwa MS, El-Emmawie AH, Saeed MA. Ocular and skin manifestations in systemic pseudohypoaldosteronism. BMJ Case Rep, 2014, 2014:bcr2014203741
[20] Kawashima Sonoyama Y, Tajima T, Fujimoto M, et al. A novel frameshift mutation in NR3C2 leads to decreased expression of mineralocorticoid receptor: a family with renal pseudohypoaldosteronism type 1. Endocr J, 2017, 64(1): 83-90
[21] Kanda K, Nozu K, Yokoyama N, et al. Autosomal dominant pseudohypoaldosteronism type 1 with a novel splice site mutation in MR gene. BMC Nephrol, 2009, 10: 37
[22] Falhammar H, Nordenstrom A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine, 2015, 50(1): 32-50
[23] Bizzarri C, Pedicelli S, Cappa M, et al. Water Balance and 'Salt Wasting' in the First Year of Life: The Role of Aldosterone-Signaling Defects. Horm Res Paediatr, 2016, 86(3):143-153
[24] Santos F, Ordonez FA, Claramunt-Taberner D, et al. Clinical and laboratory approaches in the diagnosis of renal tubular acidosis. Pediatr Nephrol, 2015, 30(12):2099-2107
[25] Hanukoglu I, Hanukoglu A. Epithelial sodium channel (ENaC) family: Phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene, 2016, 579(2): 95-132
2017-09-14
2017-10-11)
(本文编辑:孙晋枫)
Multi-systempseudohypoaldosteronismtypeⅠ:Acasereportandliteraturereview
ZOULiang-yan1,4,ZENGLi-chun2,4,JIANGSi-yuan1,CHENXiang3,YANGLin3,ZHOUWen-hao3,CHENLi-ping2,WANGLai-shuan1,3
(1DepartmentofNeonatology,Children'sHospitalofFudanUniversity,Shanghai201102,China;2DepartmentofNeonatology,Children'sHospitalofJiangxiProvince,Jiangxi330006;3TheTranslationalMedicineCenterofChildrenDevelopmentandDiseaseofFudanUniversity,ShanghaiKeyLaboratoryofBirthDefects,Children'sHospitalofFudanUniversity,Shanghai201102,China;4Co-firstauthor)
WANG Lai-shuan, E-mail: laishuanwang@163.com;CHEN Li-ping, E-mail: ccllpp88@qq.com
ObjectiveTo report a neonate diagnosed as multi-system pseudohypoaldosteronism typeⅠ(PHAⅠ)caused bySCNN1Agene heterozygous mutation,provide the basis for early diagnosis and clinical decision-making of PHAⅠ.MethodsAnalysis was performed on clinical manifestation,imageological examination,parental sanger test and treatment process of a patient carrying a pair of compound heterozygous mutaions ofSCNN1A,and literatures about clinical features of PHAⅠ.ResultsA 17-day-old boy presented with electrolyte disturbances including hyponatremia,hyperkalemia, and metabolic acidosis for 6 days. The effects of sodium supplement, hydrocortisone and insulin were not satisfactory.A pair of compound heterozygous mutations ofSCNN1Awas found by WES. c.1439+1Ggt;C was from father and had reported as a pathogenic mutation of PHAⅠ by HGMD,while c.104delC(p.P35LfsTer14) was a novel deletion mutation from mother.The patient was finally diagnosed as multi-system PHAⅠ. The children's parents gave up to treat with oral potassium lowering resin, and the children died the fourth days after discharge. Databases were searched including PubMed, CNKI, Wan Fang Database, China Science and Technology Database and CBMdisc,which described the multi-system PHAⅠfrom database-buiding time to September 4,2017. A total of 14 articles (13 English, 1 Chinese) were screened out. Renal manifestations were found in all 34 cases. Except 3 cases ofSCNN1Aand 1 case ofSCNN1Gdid not mention multi-system manifestations, the remaining cases reported dehydration and respiratory changes were the most commom manifestations,followed by stunted growth and digestive manifestations.4 cases died of cardiac arrest caused by hyperkalemia, all of them wereSCNN1Amutations, except our study with heterozygous mutation the rest were homozygous mutations.ConclusionPseudohypoaldosteronism should be considered when lasted intractable hyperkalemia, hyponatremia and metabolic acidosis happened. Neonatal gene sequencing can help diagnosis.
SCNN1Agene; Pseudohypoaldosteronism; Hyperkalemia; Hyponatremia; Metabolic acidosis; Gene diagnosis
1 复旦大学附属儿科医院新生儿科 上海,201102;2 江西省儿童医院新生儿科 江西,330006;3 复旦大学附属儿科医院,上海市出生缺陷防治重点实验室,复旦大学儿童发育与疾病转化医学研究中心 上海,201102;4 共同第一作者
王来栓,E-mail:laishuanwang@163.com;陈丽萍,E-mail:ccllpp88@qq.com
10.3969/j.issn.1673-5501.2017.05.012
