PORCN基因嵌合突变致男性局灶性真皮发育不全1例并文献复习
2017-12-02徐丹丹郑章乾吴冰冰罗飞宏
徐丹丹 陆 炜 郑章乾 杨 琳 吴冰冰 罗飞宏
PORCN基因嵌合突变致男性局灶性真皮发育不全1例并文献复习
徐丹丹1)陆 炜1)郑章乾1)杨 琳1)吴冰冰2)罗飞宏1)
目的 报道1例PORCN基因嵌合突变致男性局灶性真皮发育不全(FDH)患儿并文献复习,为该病的临床诊断提供参考。方法 总结患儿的临床表现、辅助检查和基因测序结果。在Pubmed、万方数据库和中国知网中检索建库至2017年9月30日报道的PORCN突变致FDH综合征的病例,归纳该病的临床表现,筛选并总结存活男性患儿的基因型和临床表型。结果 患儿男,12岁2月,因“身材矮小”就诊。当地医院检查胰岛素样生长因子1(IGF1)343.8 ng·mL-1,胰岛素样生长因子结合蛋白3(IGFBP3)4.9 μg·mL-1;垂体MR增强扫描未见异常;B超检查双侧睾丸、肾上腺未见异常。身高142cm(-1.4 SD),体重36.1 kg;左侧第4脚趾明显小于右侧;左侧腹部和腿部有沿Blaschko线的色素减退,左侧臀部及阴茎左侧面有浅黄色脂肪膨出;双足X线正位片示,左足第1、4、5跖骨和左拇趾第1趾骨较细,第4趾骨细小,左拇趾末节趾骨短、末端细尖。性激素6项未见异常。韦氏儿童智力量表测试显示语言、总分分值偏低。基因检测显示PORCN基因c.178Ggt;A嵌合突变,确诊为PORCN基因嵌合突变致FDH。共检索到36篇英文文献报道了经基因检测确诊为PORCN突变导致的FDH综合征205例,其中男性22例(3例在出生后死亡);临床表现以皮肤(72.7%)、骨骼系统(66.8%)和颅面部(58.5%)最常见。20例(包括本文1例)存活的PORCN突变导致的FDH综合征男性患儿中除1例为46,XXY Klinefelter综合征外,余均为嵌合体或合子后嵌合;均存在皮肤发育不全,其他临床表现多样。结论 FDH不仅可表现为肢体和皮肤异常,还可导致智力发育迟滞。PORCN基因突变所致FDH为X连锁显性遗传病,男性杂合患者多为胚胎致死性,存活男性多为嵌合突变且临床表现异质性高,临床易漏诊,对存在皮肤相似病变怀疑该病者应做基因检测以辅助诊断。
局灶性真皮发育不全;PORCN; 遗传特性; 临床表现
1 病例资料
患儿男,12岁2月,因“身材矮小”于2016年9月14日至复旦大学附属儿科医院(我院)就诊。
家长诉患儿生后6岁起较同龄人身材矮小,生长缓慢,挑食,无头痛、视物模糊、胸闷和乏力等,未予治疗。2016年8月当地医院行垂体MR增强扫描未见明显异常;B超显示双侧睾丸、肾上腺未见明显异常;胰岛素样生长因子1(IGF1)343.8 ng·mL-1,胰岛素样生长因子结合蛋白 3(IGFBP3)4.9 μg·mL-1。未有明确诊断遂至我院就诊。
患儿系G2P1,足月顺产,出生体重3.5 kg。父母非近亲结婚,父身高173 cm,母身高156 cm,无家族矮小病史。
体格检查:身高142 cm(-1.4 SD),体重36.1 kg。左侧第4脚趾明显小于右侧,双手掌大小和皮肤颜色基本正常。图1 A~C显示,左侧腹部和腿部出现沿Blaschko线的色素减退,左侧臀部及阴茎左侧面有浅黄色脂肪膨出。患儿心肺听诊,口、眼和耳等检查未见异常,神经反射正常。
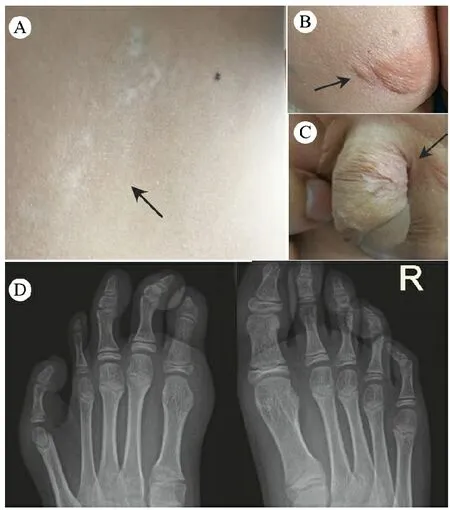
图1患儿皮肤和影像学表现
注 A:左侧腹部沿Blaschko线分布的色素缺失;B、C:臀部和阴茎有浅黄色脂肪膨出;D:X线片显示,左足第1、4、5跖骨和左拇趾第1趾骨较细,第4趾骨细小,左拇趾末节趾骨短、末端细尖卵泡刺激素(FSH)2.16 U·L-1,人绒毛膜促性腺激素(HCG)0.15 U·L-1,黄体生成素(LH)2.54 U·L-1,催乳素(PRL)9.13 nmol·L-1,雄烯二酮(AND)1.64 nmol·L-1,睾酮(TES)99.5 ng·dL-1。
实验室检查:雌二醇(E2)25 pmol·L-1,影像学检查:图2双足X线正位片示,左足第1、4、5跖骨和左拇趾第1趾骨较细,第4趾骨细小,左拇趾末节趾骨短、末端细尖。
韦氏儿童智力量表(WISC-R)测试结果总分74,其中语言总分73,操作总分83。
取得患儿父母知情同意后行全外显子测序(WES)。依据我院分子诊断中心数据分析流程,结合WuXi Next CODE分析软件(CSE)分析测序数据;通过Burrow-Wheeler Aligner (BWA)与NCBI RefSeq进行匹配比对;采用ANNOVAR和VEP软件以及注释程序注释变异数据,包括用NCBI RefSeq和SwissPort进行基因注释,用HGMD、OMIM和ClinVar进行疾病相关注释,用千人基因组计划、EVC6500、ExAC和我院分子诊断中心已建立的内部数据库进行突变频率注释;用SIFT、Polyphen 2和MutationTaster进行突变预测。通过突变频率和变异类别的筛选及与疾病的相关关系筛选出候选突变。
患儿PORCN基因(NM_203475)3号外显子存在1个杂合突变c.178Ggt;A(p.G60R),为嵌合变异(正常读序为140,突变读序为31,变异嵌合比例18%)。PORCN(MIM:305600)是X连锁显性遗传的局灶性真皮发育不全(FDH)的致病基因。对患儿及其父母进行Sanger验证,目的条带送深圳华大基因股份有限公司测序(PORCN基因PCR引物F:TCTTGTTCTCCCTCTCTCTCTC,R:CTGTGTCTCCTCATAGGCAAAT)。反向测序(图2)可见患儿该位点出现一稍低的突变峰,其父母均未出现该位点的突变峰。
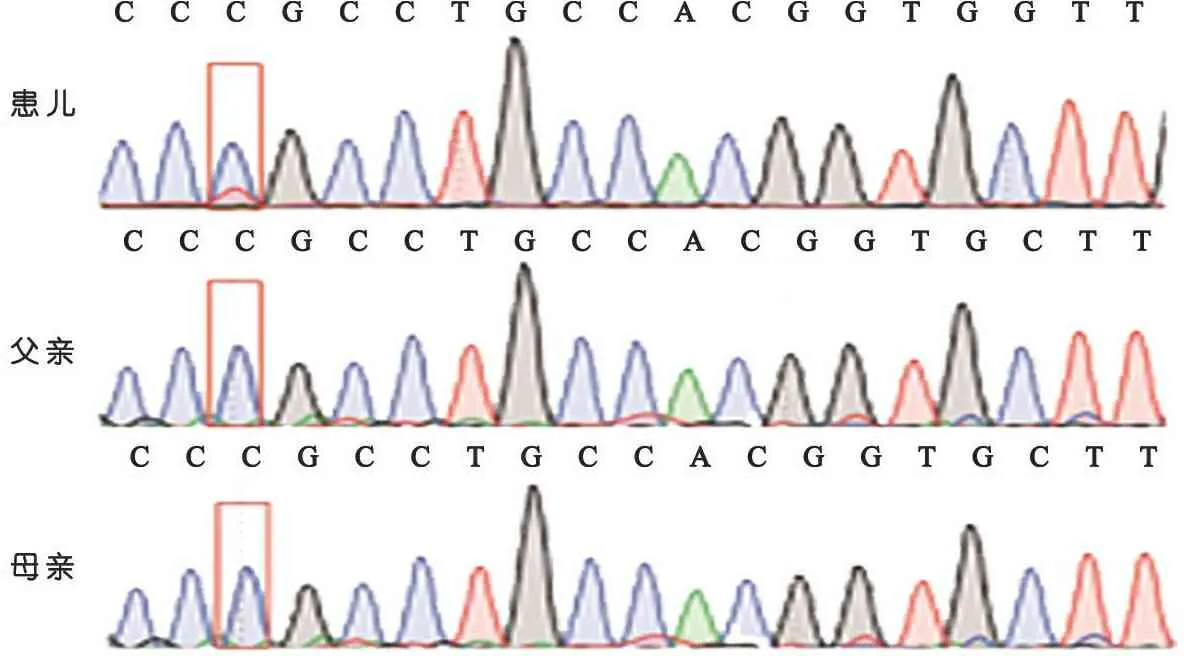
图2患儿及其父母测序图谱
根据基因测序结果结合临床诊断为PORCN基因突变导致的FDH。根据家长意愿予GnRH抑制过早发育,以延长其身高增长时间。随访至2017年7月身高146 cm(-1.7 SD),停止用药,体重36.5 kg,骨龄11.5岁。
2 文献复习
以“Focal dermal hypoplasia”、“Goltz sydrome”、“PORCN”为关键词在Pubmed数据库检索,以“局灶性真皮发育不全”、“Goltz综合征”、“PORCN”为关键词在万方数据库和中国知网检索,检索时间从建库至2017年9月30日。共检索到FDH和PORCN突变相关文献390篇,逐篇筛选后,36篇英文文献报道了经基因检测确诊为PORCN突变导致的FDH 205例,来自北美洲、欧洲和亚洲等,涉及184种突变类型,其中165例、170种突变已包含于LOVD(Leiden Open Variation Database)数据库。205例中男性患儿22例,3例生后死亡。
表1总结了文献报道的205例并本文1例PORCN突变导致的FDH的临床表现。皮肤(72.7%)、骨骼系统(66.8%)和颅面部(58.5%)表现最常见,皮肤异常主要表现为沿Blaschko线分布的皮肤色素沉着过度或减退,骨骼系统的主要表现为并指(趾)、缺指(趾),此外,面部不对称、耳朵形态异常、指甲发育不全、多部位刺瘤、头发块状缺失、牙齿异常(少牙畸形、牙釉质发育不全)、唇腭裂、眼睛异常(小眼,无眼,虹膜、脉络膜、视网膜缺损)等也在多例患儿中出现,腹股沟疝、隐睾、甲状腺肿瘤、白内障等仅在个别病例出现(未列在表格中)。表2总结了已报道的19例存活男性患儿的临床症状和突变类型,可见嵌合突变患儿的临床症状仍以皮肤发育不全为主(84.2%),并指和指甲发育不全等也较常见。

表1 206例PORCN突变导致FDH的主要临床表现[n(%)]
3 讨论
FDH又称Goltz-Gorlin综合征,最早于1962年由Goltz和Gorlin等报道[12,13],主要特征是多发畸形和发育不良,主要影响外胚层和内胚层来源的组织器官,包括皮肤、骨骼和眼睛等。目前国内报道30余例,国外报道300余例,本文文献复习共检索到205例基因测序证实为PORCN基因突变的FDH病例。该病为X连锁显性遗传病,为PORCN基因突变导致。PORCN位于Xp11.23,编码由461个氨基酸组成的52 kDa的蛋白。PORCN为PORC蛋白家族的成员,目前推测PORC蛋白是一种O-酰基转移酶,作用于Wnt通路蛋白的棕榈酰化,Wnt通路在胚胎过程中组织发育中起重要作用。PORCN是膜结合的O-酰基转移酶,有多个跨膜区域跨过内质网,并且有1个连接到膜上的O-酰基转移酶区[14-16]。此基因编码的蛋白是涉及WNT蛋白如WNT7A等合成过程中的内质网跨膜蛋白。该基因突变在男性常为致死性,目前报道的病例中男女比例约为1∶9,存活的男性患者常常为合子后体细胞突变的嵌合。
FDH为多系统疾病,皮肤、骨骼系统、面部、眼睛、口腔等均可受累,最初被报道时主要有皮肤、指/趾和眼相关临床表现[12,13]随着患者例数的增多,多种其他临床表现也相继被报道[1,17-20],该病的诊断需具备至少3种典型的外胚层相关组织器官改变及1种典型的肢体畸形,PORCN基因检测有助于确诊[3,10]。治疗主要以对症治疗为主,对可能引起感染的皮肤症状进行治疗,对可能引起严重胃食管疾病的咽喉、气管、食管巨大疣样刺瘤进行手术治疗,对影响功能的并指(趾)、缺指(趾)进行功能训练及物理治疗,对截断面缺陷的患者进行假体移植等,并定期随访。
PORCN为X连锁遗传,女性患儿多为PORCN的杂合或嵌合突变;出现在男性的半合突变多为胚胎致死性,因此存活的男性患儿基本上为嵌合体或合子后嵌合。本文检索到的205例中男性患儿22例,3例生后死亡。Madan等[21]报告了1例生后42 d死亡的杂合患儿,Brady等[22]报告了2例分别在生后10 d死亡的杂合男性患儿。表2总结了文献19例并本文1例共20例存活的PORCN突变导致的FDH男性患儿的基因型和临床表型。除例14为46,XXY Klinefelter综合征[7]外,其余均为嵌合体或合子后嵌合;PORCN突变类型包括无义突变、移码突变、错义突变等;可能受嵌合比例或突变类型的影响而临床表现有所不同,但均存在皮肤发育不全,符合FDH的诊断。
目前国内无PORCN导致男性FDH的报道,仅有2例PORCN错义突变和移码突变的女性患儿报道[23],c.1186cgt;T为已报道的突变,c.808-811delGGGG为新发突变。本文报道国内首例男性PORCN突变患儿,该突变位点已收录于HGMD数据库中,以臀部和阴茎皮肤出现浅黄色斑片、腹部及双下肢出现皮肤沿Blaschko线分布的线性色素缺失、身材矮小为主要特征,其他症状包括左侧第四趾较小,WES显示该患儿存在嵌合比例约为18%的PORCN突变c.178Ggt;A,为错义突变。患儿的临床表现和基因检查结果均符合FDH诊断标准。该病尚无有效的治疗措施,因此良好的遗传咨询及植入前遗传学诊断非常重要。母方为PORCN杂合致病患者有50%可能遗传给下一代,但由于男性杂合子常无法存活,因此其后代为33%正常女性,33%患病女性,33%正常男性;亲代若为嵌合患者,则遗传给子代的比例与嵌合比例及合子情况决定,也可能高达50%。PORCN突变类型有多种,突变类型不同也导致临床表现的严重程度不一,在LOVD数据库中以无义突变、错义突变及碱基替换为主。
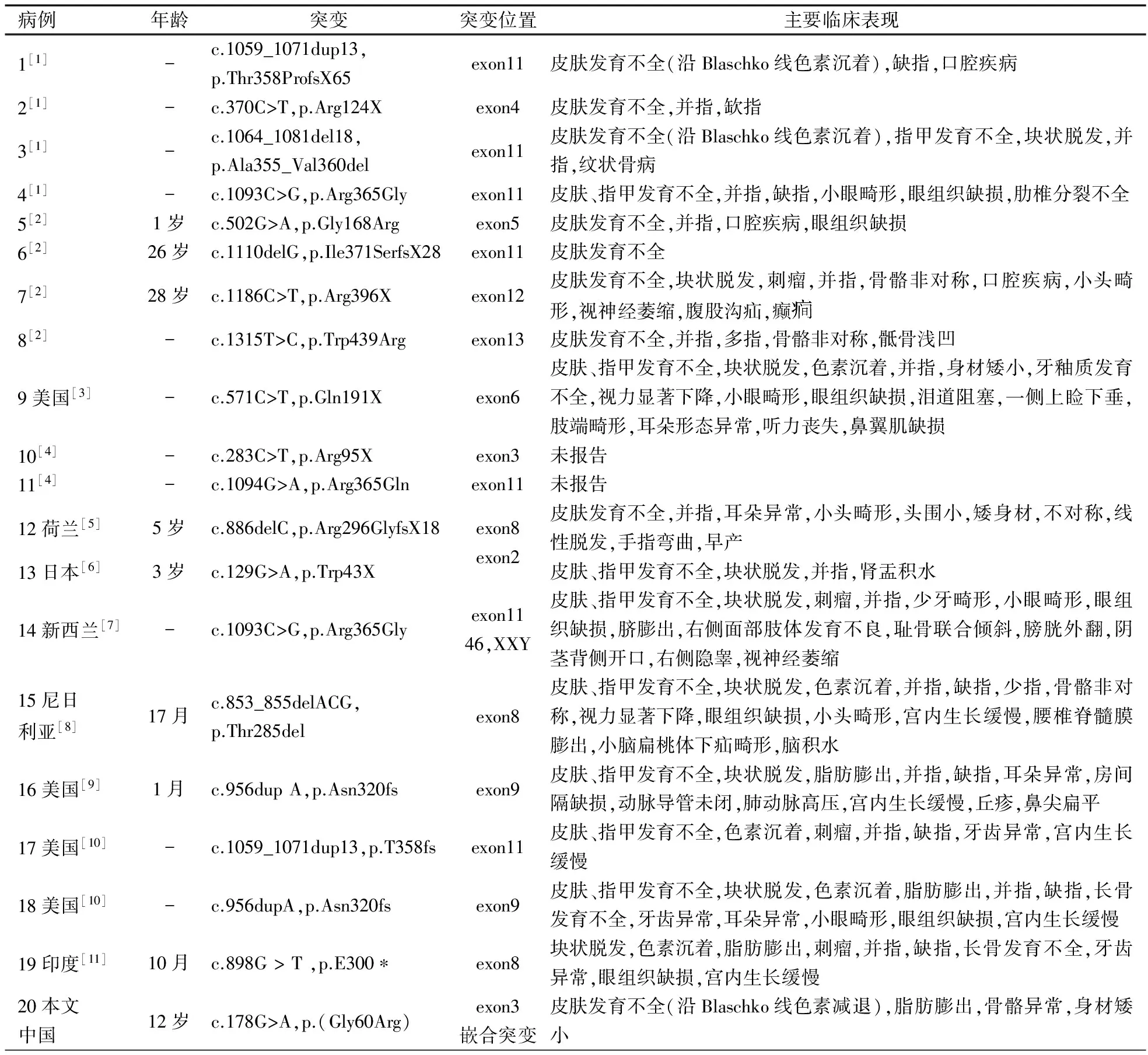
表2 20例男性PORCN突变患儿基因型和临床表型
注 -:不详
[1]Wang X, Reid Sutton V, Omar Peraza-Llanes J, et al. Mutations in X-linked PORCN, a putative regulator of Wnt signaling, cause focal dermal hypoplasia. Nat Genet, 2007, 39(7): 836-838
[2]Bornholdt D, Oeffner F, König A, et al. PORCN mutations in focal dermal hypoplasia: coping with lethality. Hum Mutat, 2009, 30(5): E618-628
[3]Maas SM, Lombardi MP, van Essen AJ, et al. Phenotype and genotype in 17 patients with Goltz-Gorlin syndrome. J Med Genet, 2009, 46(10): 716-720
[4]Fernandes PH, Wen S, Sutton VR, et al. PORCN mutations and variants identified in patients with focal dermal hypoplasia through diagnostic gene sequencing. Genet Test Mol Biomarkers, 2010, 14(5): 709-713
[5]Vreeburg M, van Geel M, van den Heuij LG, et al. Focal dermal hypoplasia in a male patient due to mosaicism for a novel PORCN single nucleotide deletion. J Eur Acad Dermatol Venereol, 2011, 25(5): 592-595
[6]Yoshihashi H, Ohki H, Torii C, et al. Survival of a male mosaic for PORCN mutation with mild focal dermal hypoplasia phenotype. Pediatr Dermatol, 2011, 28(5): 550-554
[7]Alkindi S, Battin M, Aftimos S, et al. Focal dermal hypoplasia due to a novel mutation in a boy with Klinefelter syndrome. Pediatr Dermatol, 2013, 30(4): 476-479
[8]Peters T, Perrier R, Haber RM. Focal dermal hypoplasia: report of a case with myelomeningocele, Arnold-Chiari malformation and hydrocephalus with a review of neurologic manifestations of Goltz syndrome. Pediatr Dermatol, 2014, 31 (2): 220-224
[9]Stevenson DA, Chirpich M, Contreras Y, et al. Goltz syndrome and PORCN mosaicism. Int J Dermatol, 2014, 53(12): 1481-1484
[10]Bostwick B, Fang P, Patel A, et al. Phenotypic and molecular characterization of focal dermal hypoplasia in 18 individuals. Am J Med Genet C Semin Med Genet, 2016, 172C(1): 9-20
[11]Rao SS, Shenoy RD, Salian S, et al. Focal Dermal Hypoplasia with a De novo Mutation p. E300* of PORCN Gene in a Male Infant. Indian J Dermatol, 2016, 61(6): 700
[12]Goltz RW, Peterson WC, Gorlin RJ, et al. Focal dermal hypoplasia. Arch Dermatol, 1962, 86: 708-717
[13]Goltz RW. Focal dermal hypoplasia syndrome. An update. Arch Dermatol, 1992, 128(8): 1108-1111
[14]Grigoryan T, Wend P, Klaus A, et al. Deciphering the function of canonical Wnt signals in development and disease: conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev, 2008, 22(17): 2308-2341
[15]Adaimy L, Chouery E, Megarbane H, et al. Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: the odonto-onycho-dermal dysplasia. Am J Hum Genet, 2007, 81(4): 821-828
[16]Grzeschik KH, Bornholdt D, Oeffner F, et al. Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat Genet, 2007, 39(7): 833-835
[17]Smith A, Hunt TR 3rd. The orthopedic characterization of Goltz syndrome. Am J Med Genet C Semin Med Genet, 2016, 172C(1): 41-43
[18]Bree AF, Grange DK, Hicks MJ, et al. Dermatologic findings of focal dermal hypoplasia (Goltz syndrome). Am J Med Genet C Semin Med Genet, 2016, 172C(1): 44-51
[19]Wright JT, Puranik CP, Farrington F. Oral phenotype and variation in focal dermal hypoplasia. Am J Med Genet C Semin Med Genet, 2016, 172C(1): 52-58
[20]Gisseman JD, Herce HH. Ophthalmologic manifestations of focal dermal hypoplasia (Goltz syndrome): A case series of 18 patients. Am J Med Genet C Semin Med Genet, 2016, 172C(1): 59-63
[21]Madan S, Liu W, Lu JT, et al. A non-mosaic PORCN mutation in a male with severe congenital anomalies overlapping focal dermal hypoplasia. Mol Genet Metab Rep, 2017, 12: 57-61
[22]Brady PD, Van Esch H, Fieremans N, et al. Expanding the phenotypic spectrum of PORCN variants in two males with syndromic microphthalmia. Eur J Hum Genet, 2015, 23(4): 551-554
[23]Liang Y, Liu YX, Xu ZG, et al. Two female cases of focal dermal hypoplasia: One new case with a novel variant in PORCN (c. 808_811delGGGG). J Dermatol, 2016, doi: 10.1111/1346-8138.13737
2017-10-12
2017-10-20)
(本文编辑:孙晋枫)
PORCNgenemosaicmutationcausefocaldermalhypoplasia:Acasereportandliteraturereview
XUDan-dan1),LUWei1),ZHENGZhang-qian1),YANGLin1),WUBing-bing2),LUOFei-hong1)
(Children'sHospitalofFudanUniversity,Shanghai, 201102,China; 1)DepartmentofPediatricEndocrinologyandInheritedMetabolicDisease, 2)TheMolecularGeneticDiagnosisCenter,ShanghaiKeyLabofBirthDefect)
LUO Fei-hong, E-mail: luofh@fudan.edu.cn; WU Bing-bing, E-mail: bingbingwu2010@163.com
ObjectiveTo report one case of male focal dermal hypoplasia (FDH) caused by mosaic mutation ofPORCNfor clinical diagnosis reference.MethodsThe clinical manifestations, laboratory measurements and gene sequencing results were summarized.PORCNmutations from Pubmed, Wanfang Database and China National Knowledge Infrastructure up to September 30th, 2017 were searched, the related features along with the clinical and gene mutation spectrums of the survived FDH male cases were summarized.ResultsA 12 years and 2 months old boy with height 142 cm (-1.4 SD) and weight 36.1 kg was referred to our clinic due to short stature. The serum IGF1 level was 323.8 ng·mL-1and IGFBP3 level was 4.9 μg·mL-1.No abnormalities were found in his pituitary and bilateral testes and adrenal gland. However, typical features were found as short left fourth toe, skin hypopigmentation at his left leg and abdomen along Blaschko line, and fat herniation in buttocks and penis. Bipedal X-ray showed that the 1st, 4th, 5th metatarsal of left foot, left great toe and the 4th phalanges were small, the phalanges of left great toe were short with small end. Six steroid sex hormones were within normal range. WISC-R intellectual test demonstrated that language and total scores are marginally low. Mosaic c.178Ggt; A mutation ofPORCNgene was detected by whole exon sequencing and confirmed by Sanger sequencing, then the diagnosis of FDH was made. A total of 205 reported patients confirmed havingPORCNmutations were reported in 36 literatures and only 22 were males(3 cases died soon after birth). The most frequently reported symptoms were skin hypoplasia (72.7%),skeletonabnormalities (66.8%) and craniofacial anomalies (58.5%). The survived male patients were all mosaic or postzygotic mosaic mutations except one 46, XXY Klinefelter syndrome whose clinical manifestations showed great heterogeneity but all with skin hypoplasia.ConclusionThe FDH patient we reported not only presented with limb and skin abnormalities, but also with mental retardation. Male patients could be misdiagnosed by Sanger sequencing because of the failure of detection of mosaic mutation and great heterogeneity of clinical manifestations.WES test could significantly improve the positive rate of diagnosis for those suspected male patients.
Focal dermal hypoplasia;PORCN; Genetic features; Clinical manifestation
复旦大学附属儿科医院 上海,201102; 1) 内分泌遗传代谢科;2) 分子诊断中心,上海市出生缺陷防治重点实验室
罗飞宏,E-mail:luofh@fudan.edu.cn; 吴冰冰,E-mail:bingbingwu2010@163.com
10.3969/j.issn.1673-5501.2017.05.011
