蔬菜、茶叶中克百威酶联免疫检测方法的建立
2017-11-30何方洋冯才伟杜美红王建霞桑旭升冯静
何方洋+冯才伟+杜美红+王建霞+桑旭升+冯静

摘要:为建立快速检测蔬菜、茶叶中克百威残留的酶联免疫方法,合成克百威半抗原,并与载体蛋白偶联合成人工抗原,然后免疫制备其单克隆抗体,在筛选克百威单克隆抗体的基础上,建立酶联免疫检测方法。结果表明,Logit/Log拟合标准曲线为y=-2.054x+0.667 3,相关系数(r)为0.998 5,半数抑制浓度(IC50)为2.3 μg/L,对蔬菜、茶叶样本的检测限分别为10、5 μg/kg,加标回收率为79.2%~102.7%,样本重复检测的变异系数为5.5%~11.5%。结果表明,建立的酶联免疫检测方法具有较好的特异性、准确性和重复稳定性,可用于蔬菜、茶叶中克百威残留的检测。
关键词:酶联免疫法;克百威;蔬菜;茶叶;载体蛋白;单克隆抗体;半数抑制浓度;加标回收率残留;检测
中图分类号: TS207.5+3 文献标志码: A 文章编号:1002-1302(2017)20-0213-03
克百威商品名呋喃丹,是一种广谱、高效、药效残留期长的内吸性杀虫、杀螨、杀线虫剂,广泛应用于蔬菜、水果和粮食作物等的害虫防治中[1]。因克百威对人和动物有很高的毒性,并且不易降解,容易造成环境污染,我国已规定食品中克百威的最大残留限量,但违规使用现象仍然较多,因此加强对克百威农药残留的检测十分必要[2-5]。
目前,克百威殘留分析一般使用气相色谱法(简称GC)[6]、高效液相色谱法(简称HPLC)[7-8]及色谱质谱联用技术[9-12],这些方法灵敏、准确,可以同时测定多种药物,但样品前处理复杂、繁琐费时,且需要昂贵的仪器设备和专业的操作人员,检测成本高,难以满足样品现场、批量、快速检测的需要。因此,开发一种简单快速、适用于农药残留现场监控的分析方法具有重要意义。本研究基于酶联免疫吸附技术,建立了一种快速、灵敏的检测蔬菜、茶叶中克百威残留的方法。
1 材料与方法
1.1 材料与试剂
雌性Balb/c小鼠(6~8周龄),清洁级,由北京勤邦生物技术有限公司实验动物室提供;呋喃酚、碳酰氯、克百威(纯度≥99%)、牛血清白蛋白(简称BSA)、卵清蛋白(简称OVA)均购自Sigma公司;其他化学试剂为国产分析纯;样本提取剂:称取4.40 g十二水合磷酸氢二钠和13.68 g二水合磷酸二氢钠,加入500 mL去离子水,溶解混匀。
1.2 仪器与设备
8010S匀浆机(上海斯伯明仪器设备有限公司);2000SBL电子天平(美国Setra公司);KS-Ⅱ振荡器(上海跃进医疗器械厂);QL-901漩涡混合器(江苏省海门市其林贝尔仪器制造有限公司);Anke TDL-40B低速离心机(上海安亭科学仪器有限公司);微量移液器(单道20~200、100~1 000 μL,多道20~300 μL,美国Thermo公司);DHP-600生化培养箱(天津市中环实验电炉有限公司);MK3酶标仪(美国Thermo公司)。
1.3 方法
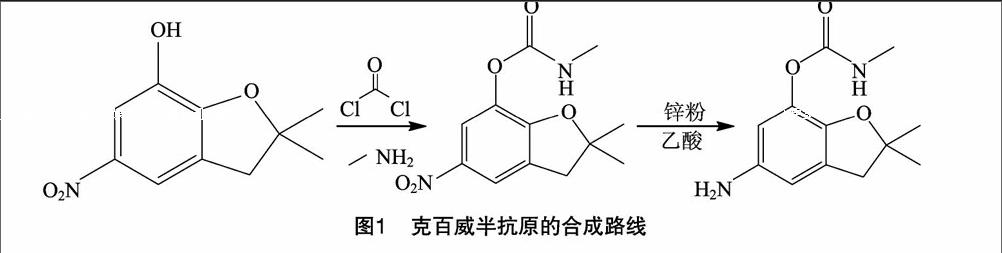
1.3.1 克百威半抗原的合成 合成路线见图1。取呋喃酚1.0 g,加30 mL二氯甲烷溶解,加入碳酰氯,0~5 ℃反应4 h,加入90 mL石油醚,静置,底部有油状物析出,转入分液漏斗中,分去上层有机相,油状物加四氢呋喃溶解,然后加入2 mL含有甲胺的四氢呋喃溶液,0~5 ℃反应2 h。停止反应,旋蒸,除去四氢呋喃,得到红色油状物,上硅胶柱,石油醚-乙酸乙酯(20 ∶ 1,体积分数)洗脱分离,得到产物Ⅰ。取锌粉 1.2 g,加蒸馏水和0.5 mL冰乙酸,80 ℃加热活化30 min,加入含有产物Ⅰ的乙醇30 mL,60 ℃反应4 h,TCL监测,反应已完全,停止反应,过滤,除去锌粉。乙醇水溶液蒸干,加水-乙酸乙酯萃取分离,分出有机相水洗干燥,石油醚-乙酸乙酯(30 ∶ 1,体积分数)重结晶,即得到克百威半抗原产物。其化学结构用核磁共振鉴定。
1.3.2 人工抗原的制备 采用碳二亚胺法[13],将克百威半抗原与牛血清白蛋白和卵清蛋白分别偶联,作为免疫原和包被原。采用2,4,6-三硝基苯磺酸(TNBS)法[14-15]测定半抗原与载体蛋白的偶联比。
1.3.3 单克隆抗体的制备 按照常规方法免疫Balb/c小鼠制备腹水抗体,用饱和硫酸铵法纯化后备用[13]。
1.3.4 间接竞争ELISA方法的建立 将包被原包被在96孔酶标板上,100 μL/孔,37 ℃温育2 h;倒去板中溶液,经PBST洗涤3次,加入封闭液200 μL/孔,37 ℃温育2 h;洗涤3次,加入系列浓度的克百威标准溶液50 μL/孔和单克隆抗体溶液50 μL/孔,25 ℃避光反应30 min;洗涤4~5次后,加入酶标二抗100 μL/孔,25 ℃避光反应30 min; 洗涤4~5次后加入底物液A液(过氧化脲)和B液(四甲基联苯胺)各 50 μL/孔,25 ℃显色15 min,加入2 mol/L H2SO4终止液 50 μL/孔,设定酶标仪于450 nm处测定每孔吸光度。
1.3.5 标准曲线的绘制 采用间接竞争ELISA方法建立标准曲线,分别选择克百威标准品浓度0.0、0.5、1.5、4.5、13.5、40.5 μg/L,以浓度为0.0 μg/L时的吸光度为D0值,相应克百威浓度的吸光度为D值,以百分吸光度(D/D0)为纵坐标,标准品浓度的对数值为横坐标,绘制标准曲线。
1.3.6 样本前处理方法的建立
1.3.6.1 蔬菜前处理方法 称取(1.00±0.05)g均质后的蔬菜样品至10 mL聚苯乙烯离心管中,加入5 mL样本提取剂,用振荡器振荡5 min,混匀,4 000 r/min室温离心5 min;移取 200 μL 上清液至2 mL聚苯乙烯离心管中,加入600 μL样本复溶工作液,用涡旋仪涡动2 min,充分混匀;取50 μL用于分析。endprint
1.3.6.2 茶叶前处理方法 称取1 g茶叶样本至50 mL聚苯乙烯离心管中,加入10 mL乙腈,用涡旋仪涡动1 min,混匀,4 000 r/min室温离心5 min;移取1 mL上层有机相至 10 mL 洁净干燥的玻璃管中,于20~30 ℃水浴氮气流下吹干;加入1 mL样本复溶工作液,用涡旋仪涡动2 min,充分混匀;取50 μL用于分析。
1.3.7 ELISA方法技术性能的评价
1.3.7.1 敏感性试验 用IC50和检测限来评价方法的灵敏度。测定20次标准曲线的IC50,统计其平均值和浮动范围;测定20个空白样本,求出其D/D0在标准曲线上对应浓度的平均值(x)和标准差(s),方法检测限MDL=x+3s。
1.3.7.2 准确性和重复性试验 用回收率和变异系数分别评价ELISA方法的准确性和重复性。以3个不同浓度的克百威标准品分别对空白马铃薯、黄瓜、白菜、茶叶样本进行添加回收试验,计算回收率和变异系数。
1.3.7.3 特异性试验 选择与克百威具有类似结构和类似功能的其他农药进行交叉反应性检测。按下式计算交叉反应率:交叉反应率=x/y×100%,式中x为引起50%抑制的克百威浓度,y为引起50%抑制的其他农药浓度。
2 结果与分析
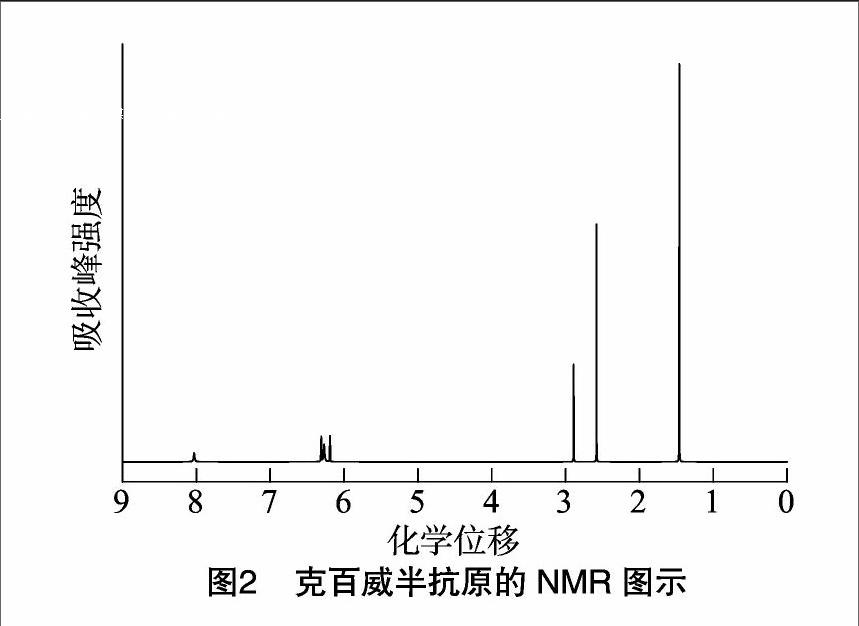
2.1 半抗原的核磁共振氫谱鉴定
取半抗原产物经核磁共振(nuclear magnetic resonance,简称NMR)得图2。从图2可得,1H-NMR(CDCl3,300 MHz)δ:8.0(s,1H,—NH—),6.27(s,2H,—NH2),6.19(s,1H,ArH),6.31(s,1H,ArH),2.89(s,2H,—CH2—),2.58(s,3H,—CH3),1.46(s,6H,—CH3)。图谱中δ=6.27的苯环上芳香胺特征吸收峰的存在,结合其他共振吸收峰的特征,证明克百威半抗原结构正确。
2.2 半抗原与载体蛋白偶联比的测定
由图3得曲线公式为y=5.174 2x3-1.554 5x2+0.621 8x-0.004 8,根据公式得BSA抗原(免疫原)的偶联比=335×(y2-y1),OVA抗原(包被原)的偶联比=225×(y2-y1)。式中:y2为载体蛋白的氨基浓度,y1为偶联抗原的氨基浓度。
结果显示,BSA抗原(免疫原)测定出的吸光度为0.149,BSA测定出的吸光度为0.205,根据公式计算出y1=0.070 452 735,y2=0.101 917 517,偶联比为335×(y2-y1)=335×(0.101 917 517-0.070 452 735)=10.54;OVA抗原(包被原)测定出的吸光度为0.121,OVA测定出的吸光度为 0.184,根据公式计算出y1=0.056 844 776,y2=0.089 214 748,偶联比为225×(y2-y1)=225×(0.089 214 748-0.056 844 776)=7.28。
2.3 标准曲线的建立
以百分吸光度(D/D0)为纵坐标(y)、标准品浓度的对数值为横坐标(x)绘制标准曲线,结果见图4。以Logit(D/D0)为纵坐标,标准品浓度的对数值为横坐标,将图4转换成图5后可知,在 0.5~40.5 μg/L浓度范围内,Logit(D/D0)与克百威浓度对数呈良好的线性关系,建立拟合回归直线方程为 y=-2.054 0x+0.667 3,相关系数r为0.998 5,半数抑制浓度(IC50)为2.3 μg/L。
2.4 敏感性试验
统计20次标准曲线的IC50平均值为2.2 μg/L,浮动范围为1.5~3.0 μg/L;20个空白样本的D/D0在标准曲线上对应浓度的平均值(x)和标准差(s)见表1。综合考虑几种样本的检测限数据,本方法对蔬菜样本的检测限确定为5 μg/kg,对茶叶样本的检测限确定为10 μg/kg。
2.5 准确性和重复性试验
取空白马铃薯、黄瓜、白菜、茶叶样本,按表2所述的克百威添加浓度对其进行添加回收试验,每个浓度做5个平行,用3个批次的试剂测定,分别计算回收率和批内、批间变异系数,结果见表2。由表2可知,以3个不同浓度的克百威标准品对空白马铃薯、黄瓜、白菜、茶叶样本进行添加,其添加回收率为79.2%~102.7%,批内变异系数为5.5%~8.1%,批间变异系数为7.7%~11.5%,均小于15%,说明用该ELISA方法测定蔬菜、茶叶中克百威的残留具有准确性和重复性,可用于蔬菜、茶叶中克百威的残留测定。
2.6 特异性试验
选择氨基甲酸酯类农药(丁硫克百威、甲萘威、异丙威、速灭威、灭多威、仲丁威)与有机磷类农药(甲胺磷、久效磷、敌敌畏)进行交叉反应性检测。结果发现, 选用的克百威单克隆抗体与丁硫克百威有一定的交叉反应(11.8%),这可能是因为克百威作为丁硫克百威的代谢产物,有类似的结构,而对其他农药的交叉反应率均较低(<1%),表明本方法具有良好的特异性。
3 结论与讨论
酶联免疫法因灵敏度高、特异性好、操作简便、检测成本低、一次性检测样本量大等特点,能够更好地满足我国食品企业、政府监管部门等开展检测工作。本研究初步建立了蔬菜、茶叶中克百威残留检测的酶联免疫吸附法。该方法的IC50浮动范围为1.5~3.0 μg/L,对蔬菜样本的检测限为5 μg/kg,对茶叶样本的检测限为10 μg/kg;样本添加回收率为 79.2%~102.7%,重复检测的变异系数为5.5%~11.5%,并且样本前处理简单、仪器设备投资少、检测成本低,适用于大批量样本中克百威残留的快速筛选。
参考文献:
[1]上海交通大学. 克百威农药的纳米胶体金标记免疫测定方法:ZL03116692.X[P]. 2003-04-29.endprint
[2]杨金易,吴 青,王 弘,等. 高亲和力的农药克百威单克隆抗体的制备及鉴定[J]. 中国农业科学,2007,40(3):518-523.
[3]刘乾开,朱国念. 新编农药使用手册[M]. 上海:上海科学出版社,1999:101-102.
[4]食品安全国家标准 食品中农药最大残留限量:GB 2763—2014[S]. 北京:中国标准出版社,2014.
[5]刘 刚. 山东省:毒死蜱氧乐果克百威在蔬菜上违规使用现象仍然较多[J]. 农药市场信息,2015(25):16.
[6]孙盈盈. 气相色谱法检测水产品中呋喃丹的残留[D]. 武汉:华中农业大学,2013.
[7]Vera-Avila L E,Márquez-Lira B P,Villanueva M,et al. Determination of carbofuran in surface water and biological tissue by sol-gel immunoaffinity extraction and on-line preconcentration/HPLC/UV analysis[J]. Talanta,2012,88:553-560.
[8]韦林洪,刘曙照,邵秀金. 免疫亲和色谱-高效液相色谱法测定河水和土壤中克百威、三唑磷和绿磺隆残留量[J]. 理化检验(化学分册),2012,48(8):887-892.
[9]张慧丽,王建华,李 馨,等. 气相色谱串联质谱法测定蔬菜中甲拌磷和克百威及其代谢物的残留量[J]. 化学分析计量,2016,25(1):10-14.
[10]Chowdhury M A Z,Fakhruddin A N M,Islam M N,et al. Detection of the residues of nineteen pesticides in fresh vegetable samples using gas chromatography-mass spectrometry[J]. Food Control,2013,34(2):457-465.
[11]張 璇,姜 敏,刘 峰,等. 高效液相色谱-质谱联用仪检测苹果中的丁硫克百威和克百威的残留量[J]. 农药科学与管理,2015,36(6):44-48.
[12]Moreno-González D,Huertas-Pérez J F,García-Campa nˇa A M,et al. Determination of carbamates in edible vegetable oils by ultra-high performance liquid chromatography-tandem mass spectrometry using a new clean-up based on zirconia for QuEChERS methodology[J]. Talanta,2014,128:299-304.
[13]杨利国,胡永昶,魏平华,等. 酶免疫测定技术[M]. 南京:南京大学出版社,1998:139-157,204-211,254-255.
[14]Habeeb A F S A. Determination of free amino groups in proteins by trinitrobenzenesulfonic acid[J]. Analytical Biochemistry,1966,14(3):328-336.
[15]Sashidhar R B,Capoor A K,Ramana D. Quantitation of ε-amino group using amino acids as reference standards by trinitrobenzene sulfonic acid:a simple spectrophotometric method for the estimation of hapten to carrier protein ratio[J]. Journal of Immunological Methods,1994,167(1/2):121-127.endprint
