PLA2G6基因纯合突变致婴儿神经轴索营养不良1例报告
2017-11-29卢洪珠陈燕惠
卢洪珠 陈 荣 陈燕惠
福建医科大学附属协和医院儿科 (福建福州 350001)
PLA2G6基因纯合突变致婴儿神经轴索营养不良1例报告
卢洪珠 陈 荣 陈燕惠
福建医科大学附属协和医院儿科 (福建福州 350001)
目的探讨婴儿神经轴索营养不良的临床特征和PLA2G6基因突变特点。方法回顾分析1例婴儿神经轴索营养不良患儿的临床资料。结果患儿,男,2岁,主要临床表现为行走、语言倒退,肌张力减低。血清总铁结合力稍低,天冬氨酸氨基转移酶和乳酸脱氢酶升高;肌电图检查示上下肢神经源性损害;彩色多普勒超声心动图、视频脑电图、颅脑磁共振均未见明显异常。基因检查结果提示患儿PLA2G6纯合突变c.1077G>A,其父母该位点均为杂合突变。结论PLA2G6基因突变导致婴儿神经轴索营养不良。
神经轴索营养不良;PLA2G6基因; 婴儿
婴儿神经轴索营养不良(infantile neuroaxonal dystrophy,INAD)是一种罕见的神经退行性病变,以运动和认知进行性倒退、肌张力减低为特征。INAD为常染色体隐性遗传病,与PLA2G6基因突变有关,患儿于3岁前起病,多在10岁前死亡[1,2]。由于疾病早期缺乏典型的临床表现,诊断较困难。现回顾分析1例PLA2G6基因突变的 INAD患儿临床资料和基因检测结果,以提高临床医师的认识。
1 临床资料
患儿,男,2岁。因“行走、语言倒退近1年”,于2016年12月29日入院。入院前1年发现患儿行走步态异常、速度减慢,独走及独站片刻会摔倒,并发现患儿学语缓慢,语言减少,从“yi-ya”到学“ba-ba”、“mama”发音后未再学会新的词汇。无抽搐,无纳差,无呕吐、腹泻,无黄疸、皮疹、发热等。随后患儿四肢无力更明显,双下肢为甚,逐步失去行走及语言能力,但尚可听懂简单指令和示意简单需求。患者系G3P2,足月顺产,出生史无异常,出生体质量3.0 kg,3个月抬头,咿呀发音,6个月坐,9个月爬,1岁站立和扶走。父母体健,非近亲婚配,有一15岁同胞姐姐,学习成绩良好,无相关疾病家族史。体格检查:体温 36.7 ℃,脉搏 108次/min,呼吸频率 28次/min,头围48 cm,身长82 cm,体质量10 kg。神志清楚,体型瘦小,巩膜无黄染,双瞳孔等大等圆,直径2.5 mm,对光反射灵敏,眼底正常;双肺呼吸音稍粗,未闻及干湿性啰音;心律齐,未闻及病理性杂音;腹软,肝脾肋下未触及;双下肢无水肿,四肢肌力检查不能配合,肌张力减低;腹壁反射、膝反射稍减弱,病理征未引出。实验室检查:血铜蓝蛋白、铁蛋白无异常,血清铁、未饱和铁结合力、运铁蛋白饱和度均正常,血清总铁结合力39.3 μmol/L(参考范围50~77 μmol/L);血常规检查,血小板计数375×109/L,淋巴细胞计数5.12×109/L,余未见明显异常;生化全套检查,天冬氨酸氨基转移酶(AST)107 IU/L,乳酸脱氢酶(LDH) 453 IU/L,余未见明显异常;血氨基酸、尿有机酸遗传代谢病筛查未见异常;尿、粪常规无异常。彩色多普勒超声心动图、视频脑电图、颅脑磁共振成像(MRI)、海马磁共振质子波谱均未见明显异常。双侧脑干听觉诱发电位异常。肌电图检查示上下肢神经源性损害。
经医院伦理委员会审查,患儿父母知情同意后,抽取患儿及其父母静脉血各2 mL,由广州金域医学检验中心进行基因检测。使用QIAamp Blood DNA Mini Kit提取基因组DNA,应用Primer Premier 5.0软件设计引物,ABI9700型PCR仪对PLA2G6基因所有外显子区及其邻近内含子区域进行扩增,产物纯化后通过ABI3500测序仪测序,所有测序结果进行机器及人工读图,运用软件Augustus分析,并与基因库中标准序列比较。发现患儿基因PLA2G6(NM-003560.2)Exon7:c.1077G>A,剪接突变,纯合,为已报道的致病突变,其父母该位点均检测到杂合突变 (图1)。结合临床表现,患儿最终确诊为INAD。诊断明确后,予维生素B1、左旋多巴、甲钴胺营养神经等对症支持治疗,但效果不佳。患儿现2岁5个月,失去语言能力,不能扶走。
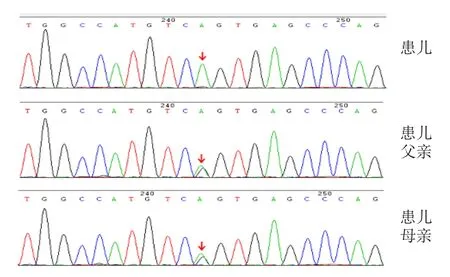
图1 患儿及其父母PLA2G6基因突变位点序列图
2 讨论
INAD为PLA2G6基因突变导致的常染色体隐性遗传神经退行性病。患儿一般在3岁前起病[2],最早可于新生儿期发病[3]。有研究曾对25例携带PLA2G6基因相关的神经退行性病的中国大陆患儿进行分析,发现平均发病年龄约15.8个月,其中24例患儿在运动能力和智力方面均快速下降,大多患儿起始表现为步态异常,但发病前,22例患儿的发育里程碑正常[4]。INAD早期,最常见临床表现为精神运动倒退,其他常见表现有肌张力降低、共济失调、眼球震颤、斜视和视神经萎缩,随着病情快速进展,继发四肢痉挛性瘫痪、认知功能障碍[5,6]。本例患儿1岁发病,发病年龄早,发病前抬头、翻身和站立等运动发育正常,起始表现为步态异常和语言减少,并且行走、语言进行性倒退,未见痉挛性瘫痪、认知障碍,可能尚处于病程早期。
实验室检查发现,本例患儿血液AST和LDH高,与其他研究报道一致[7]。有研究对31例非钙依赖型磷脂酶A2(calcium-independent phospholipase A2,iPLA2)相关性神经变性病患者的血液生化检查中发现,31例患者AST均升高,95%的患者LDH升高。AST和LDH升高考虑与PLA2G6基因编码的iPLA2 异常有关[8]。iPLA2将甘油磷脂水解为溶血磷脂和游离脂肪酸,为维持胞膜和线粒体膜稳态所必需。AST广泛存在于神经细胞中,有两种亚型,包括胞质AST和线粒体AST,线粒体AST除具有酶活性,还是细胞再摄取游离脂肪酸必需的一种细胞质膜结合蛋白。在PLA2G6基因突变的患者中,iPLA2异常,导致线粒体受损,广泛的线粒体损伤和游离脂肪酸代谢障碍可能导致受损的细胞异常释放线粒体AST,引起血液AST升高。LDH催化乳酸生成丙酮酸,几乎存在于所有组织中,组织和细胞损伤都会引起LDH升高,因此LDH升高的原因可能与弥漫性的神经损伤相关。由于AST和LDH水平升高在病程早期就可以检测到,并且会持续存在,故AST和LDH可作为潜在的iPLA2相关性神经变性病的生化标志物。本例患儿病程长达1年,加上体型瘦小,不能除外贫血、遗传性铜蓝蛋白缺乏症等这些也可能引起铁异常沉积的疾病,查血清铜蓝蛋白正常,总铁结合力稍低,血常规血小板和淋巴细胞计数稍高,但未见相关临床表现,且仅有一次检测异常,故未做处理,嘱出院后复查。可疑INAD的患者,需行脑电图和肌电图检查协助诊断。本例患儿肌电图检查示上下肢神经源性损害,视频脑电图未见明显异常,与文献报道一致[5]。
头颅MRI检查有助于INAD的诊断。研究发现,PLA2G6基因突变患儿的头颅MRI 示95%有小脑萎缩,61%可见小脑高信号,48%见苍白球区异常铁沉积,14%黑质铁增多,但2例年龄<2岁的INAD患儿头颅MRI未现异常[9]。国内对10例确诊为INAD的患儿进行随访发现,10例患儿病程1~2年后的头颅MRI均提示明显小脑萎缩[10]。本例患儿头颅MRI、MRS均未见明显异常,可能和病程尚短相关,需定期复查颅脑MRI,以便及时发现颅脑改变。
PLA2G6基因位于22q12~q13,编码iPLA2[8],参与维持膜磷脂代谢平衡和催化甘油磷脂水解。如果iPLA2缺失或功能改变,会引起膜磷脂氧化损伤后不能被修复,对膜流动性、渗透性和铁稳态产生不利影响,可能造成轴索异常,形成球状体,导致脑内铁沉积[11-13]。此外,线粒体的损伤在PLA2G6基因突变引起的神经退行性病的病理生理中发挥重要作用。研究表明,PLA2G6基因缺失后,线粒体出现异常,包括线粒体呼吸链功能障碍,ATP合成减少,线粒体形态改变[14],提示PLA2G6基因异常会导致线粒体功能紊乱和结构改变,使氧化应激增强,影响体内正常的新陈代谢。该基因突变会引起多种疾病,包括INAD/不典型INAD/脑铁沉积特发性神经变性病/Karak综合征、帕金森病[13,15],不同突变类型会造成不同的疾病,具体机制尚不清楚。PLA2G6基因突变类型有129种,包括错义突变、无义突变、插入突变、缺失突变、重复突变、框移突变、剪切位点异常。本例患儿基因PLA2G6c.1077G>A,属于剪切位点异常,即第7个外显子1077位碱基G被置换为A。该突变激活了第7个内含子5’端供体剪接位点上游的一个隐蔽剪切位点,引起iPLA2基因mRNA 在剪切过程中4个碱基的缺失(c.1074-1077 del.GTCG)和框移突变[16],使得不成熟的蛋白质截断,导致iPLA2功能改变,引起膜磷脂损伤后不能被修复,造成轴索异常、脑内铁沉积,出现行走、语言倒退,肌张力减低等表现[4,16,17]。基因PLA2G6c.1077G>A曾被报道是复合杂合突变[16]。该患儿父母此位点均为杂合突变,无相关临床症状,但患儿为纯合突变,出现异常症状。
目前国内INAD的治疗多为对症支持,患儿预后差,多在10岁前死亡[5]。早期正确诊断有助于为患儿提供更及时有效的支持治疗方式,有利于改善其生存质量。产前诊断有助于早期明确胎儿是否为携带者或患病,对已明确突变类型的家庭尤其重要,能更好指导优生优育。
综上,INAD的诊断有赖于临床表现、生化检查、头颅MRI和基因检查。精神运动倒退并进行性加重者,在婴幼儿期起病,生化检查发现AST和LDH高,应考虑INAD,需行头颅MRI检查。当MRI未见异常时,在排除其他疾病后,应及时行基因检测,检测到PLA2G6基因突变能确诊INAD,直接基因测序可发现约85%的PLA2G6突变[18]。
[1]Iodice A,Spagnoli C,Salerno GG,et al.Infantile neuroaxonal dystrophy andPLA2G6-associated neurodegeneration: an update for the diagnosis [J].Brain Dev,2017,39(2): 93-100.
[2]Sinskey JL,Holzman RS.Perioperative considerations in infantile neuroaxonal dystrophy [J].Paediatr Anaesth,2017,27(3): 322-324.
[3]Fusco C,Frattini D,Panteghini C,et al.A case of infantile neuroaxonal dystrophy of neonatal onset [J].J Child Neurol,2015,30(3): 368-370.
[4]Zhang P,Gao Z,Jiang Y,et al.Follow-up study of 25 Chinese children withPLA2G6-associated neurodegeneration [J].Eur J Neurol,2013,20(2): 322-330.
[5]Gregory A,Polster BJ,Hayflick SJ.Clinical and genetic delineation of neurodegeneration with brain iron accumulation [J].J Med Genet,2009,46(2): 73-80.
[6]Romani M,Kraoua I,Micalizzi A,et al.Infantile and childhood onsetPLA2G6-associated neurodegeneration in a large North African cohort [J].Eur J Neurol,2015,22(1):178-186.
[7]Kraoua I,Romani M,Tonduti D,et al.Elevated aspartate aminotransferase and lactate dehydrogenase levels are a constant fi nding inPLA2G6-associated neurodegeneration [J].Eur J Neurol,2016,23(4): e24-e25.
[8]Morgan NV,Westaway SK,Morton JE,et al.PLA2G6,encoding a phospholipase A2,is mutated in neurodegenerative disorders with high brain iron [J].Nat Genet,2006,38(7):752-754.
[9]Gregory A,Westaway SK,Holm IE,et al.Neurodegeneration associated with genetic defects in phospholipase A (2) [J].Neurology,2008,71(18): 1402-1409.
[10]吴晔,姜玉武,高志杰,等.婴儿神经轴索营养不良的临床、神经病理及遗传学研究.中华医学会儿科学分会.中华医学会第五次全国儿科中青年学术交流大会论文汇编(上 册) [C].中华医学会儿科学分会,2008.
[11]Balsinde J,Balboa MA.Cellular regulation and proposed biological functions of group VIA calcium-independent phospholipase A2 in activated cells [J].Cell Signal,2005,17(9): 1052-1062.
[12]Kurian MA,McNeill A,Lin JP,et al.Childhood disorders of neurodegeneration with brain iron accumulation (NBIA) [J].Dev Med Child Neurol,2011,53(5): 394-404.
[13]Salih MA,Mundwiller E,Khan AO,et al.New fi ndings in a global approach to dissect the whole phenotype ofPLA2G6gene mutations [J].PLoS One,2013,8: e76831.
[14]Kinghorn KJ,Castillo-Quan JI,Bartolome F,et al.Loss ofPLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction [J].Brain,2015,138: 1801-1816.
[15]Gui YX,Xu ZP,Wen-Lv,et al.Four novel rare mutations ofPLA2G6in Chinese population with Parkinson's disease [J].Parkinsonism Relat Disord,2013,19(1): 21-26.
[16]Lu CS,Lai SC,Wu RM,et al.PLA2G6mutations inPARK14-linked young-onset parkinsonism and sporadic Parkinson's disease [J].Am J Med Genet B Neuropsychiatr Genet,2012,159(2): 183-191.
[17]Kapoor S,Shah MH,Singh N,et al.Genetic analysis ofPLA2G6in 22 Indian families with infantile neuroaxonal dystrophy,atypical late-onset neuroaxonal dystrophy and dystonia Parkinsonism complex [J].PLoS One,2016,11(5):e0155605.
[18]Crompton D,Rehal PK,MacPherson L,et al.Multiplex ligation-dependent probe ampli fi cation (MLPA) analysis is an effective tool for the detection of novel intragenicPLA2G6mutations: implications for molecular diagnosis [J].Mol Genet Metab,2010,100 (2): 207-212.
2017-04-01)
(本文编辑:邹 强)
A homozygous mutation inPLA2G6gene causing infantile neuroaxonal dystrophy: a case report
LU Hongzhu,CHEN Rong,CHEN Yanhui (Department of Pediatrics,Union Hospital Af fi liated to Fujian Medical University,Fuzhou 350001,Fujian,China)
ObjectiveTo explore the clinical and the genetic features of infantile neuroaxonal dystrophy (INAD).MethodsThe clinical and laboratory data,neuroimaging examination and genetic testing results of one child with INAD were retrospectively analyzed.ResultsA 2 years old boy presented motor and verbal dexterity regression and hypotonia.Laboratory findings revealed decreased total iron-binding capacity in serum with increased glutamic oxaloacetic transaminase (AST) and lactic dehydrogenase (LDH).Myoelectrography showed neurogenic impairments of the arms and legs,and the color doppler ultrasound of the heart,video-EEG and brain MRI results were normal.A homozygous mutation of c.1077G>A was found inPLA2G6gene of the infant.The infant’s parents were heterozygous mutation carriers at this locus.ConclusionsPLA2G6gene mutations cause INAD.
infantile neuroaxonal dystrophy;PLA2G6gene; infant
10.3969/j.issn.1000-3606.2017.11.005
陈燕惠 电子信箱:yanhui_0655@126.com
