Cockayne 综合征1例临床及ERCC8基因突变特征
2017-11-29马秀伟赵家艳辜蕊洁封志纯
马秀伟 赵家艳 辜蕊洁 封志纯
1.陆军总医院附属八一儿童医院神经发育科(北京 100700);2.新乡医学院第三附属医院儿科(河南新乡 453000)
Cockayne 综合征1例临床及ERCC8基因突变特征
马秀伟1赵家艳2辜蕊洁1封志纯1
1.陆军总医院附属八一儿童医院神经发育科(北京 100700);2.新乡医学院第三附属医院儿科(河南新乡 453000)
目的探讨Cockayne综合征患儿的临床、影像学和ERCC8基因突变特征。方法回顾分析1例经基因检测确诊的Cockayne综合征患儿的临床和影像学资料,应用目标序列捕获和第二代测序技术检测患儿相关基因,采用Sanger测序验证突变位点的结果,并对其父母、姐姐样本进行突变位点的序列分析。结果女性患儿,7岁,主要临床表现为精神运动发育迟滞、生长发育障碍、特殊面容、光敏性皮炎、痉挛性瘫痪、小脑共济失调。头颅磁共振显示双侧半卵圆中心、脑室旁白质对称脱髓鞘改变,小脑萎缩。二代测序结果显示患儿ERCC8基因外显子区域两处杂合突变点c.397C>T和c.394_398del,分别引起氨基酸变化p.Q133X和p.L132fs;Sanger测序结果显示2个突变分别来源于母亲和父亲,为复合杂合突变。c.394_398del位点为已报道致病突变,c.397C>T为首次报道。结论二代测序技术可准确检测Cockayne综合征的ERCC8基因突变。首次发现c.397C>T突变位点,扩大了中国Cockayne综合征患者的基因突变谱。
Cockayne综合征; 临床特征;ERCC8基因; 基因突变
Cockayne综合征(Cockayne syndrome,CS;MIM#133540,216400)是一种罕见的常染色体隐性遗传病,由Cockayne在1936年首次报道,至今全世界报道200余例,欧洲报道发病率0.027/万[1],我国只有散在个例报道[2-5]。由于临床医师对CS认识不足,部分患儿可能误诊为脑性瘫痪。近年来随着目标序列捕获测序技术的应用,越来越多的神经遗传病得到明确诊断。二代测序技术具有高通量、快速、高灵敏度、自动化程度高和测序费用相对低等优势。为提高对CS的认识,现回顾分析1例经二代测序确诊的CS患儿临床表现、头颅磁共振成像(MRI)及ERCC8基因突变分析的结果。
1 临床资料
患儿,女,7岁,因“精神运动发育落后6年余”就诊于陆军总医院八一儿童医院神经发育专科门诊。患儿系G2P2,足月顺产,出生体质量3 200 g,生后无窒息。4个月能翻身,7个月可独坐,1岁时仍不能独站,不会走,不能发“baba、mama”。4个月开始皮肤容易出现皮疹,对日光敏感,暴露在日光下皮肤会发红水肿,数日后蜕皮,留下灰色色素沉着。1岁时就诊,查头颅CT、头颅MRI、骨盆正位片未见异常,18个月时行0~6岁小儿神经心理检查量表评估,单项发育商(DQ):大运动67、语言76、精细动作82、适应能力73、社交行为91。诊断脑性瘫痪,间断进行康复训练,效果不佳。5岁时复查头颅MRI显示:双侧半卵圆中心及侧脑室旁对称性异常信号,前后角旁著,小脑萎缩(图1)。视频脑电图未见异常。患儿本次就诊时7岁,能独走,协调性差,易摔跤抖动。能听懂简单指令,不能说整句话,生活不能自理。父母非近亲婚配,有一姐姐,10岁,发育正常。体格检查:消瘦,体质量15 kg,身高101cm,头围46cm。眼睛深邃,鼻细长,大耳,下颌较窄,面部散在少许灰色色素沉着,口腔见龋齿;心、肺、腹未见异常;脊柱未见畸形;四肢肌力5级,双下肢肌张力高,指鼻试验见粗大震颤,Romberg’s征阳性,膝关节有挛缩,腱反射亢进,双侧Babinski征阴性。视觉诱发电位显示:双侧P100潜伏期延长。脑干听觉诱发电位显示:双耳听通路外周段及脑干段传导延迟。
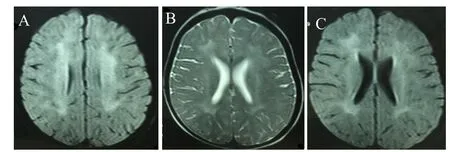
图1 患儿头颅MRI
经医院伦理委员会审核,患儿监护人签署知情同意书后,抽取患儿及其父母、姐姐外周血各2mL,应用核酸自动提取仪(百泰克AU1001)、NANODROP LITE光度计(Thermoscientific)、新型全血基因组DNA提取试剂盒(磁珠法,百泰克AU1802)提取全血基因组DNA。采用美国安捷伦公司Sure-Select 靶向序列捕获技术,委托北京金准医学检验所完成。目标区覆盖度99.6%,目标区平均深度159.92,目标区平均深度>20X比例99.2%。取1.5μg DNA应用杯式超声细胞粉碎机(新芝SCIENTZ08-III)将其随机打断,纯化后的DNA使用末端修复反应缓冲液和末端修复酶混合物进行末端补平,在其3’端加入A形成特有的悬垂结构,通过接头连接形成标准的Solexa测序文库,取1μL制备好的文库样本,用Nanodrop 2000对文库样本进行定量。文库经PCR扩增后,取3μg DNA,应用北京金准基因科技有限公司研发的神经遗传病相关基因检测试剂盒进行杂交,洗脱的杂交文库质控合格后经Hiseq2500/Hiseq2000上机测序。按照本实验常规的Sanger法对点突变患儿样本进行测序验证,并对其父母、姐姐样本进行该位点的序列分析。外显子序列由http://www.ncbi.nlm.nih.gov获得,根据软件Primer5.0设计引物。发现的新突变在人类基因突变数据库(HGMD)及千人基因组数据库(1000Genomes)查看是否已有报道,并在100名正常人中进一步验证。测序结果显示:患儿ERCC8基因外显子区域存在两处杂合突变点,chr5:60214094区域 c.397C>T,导致氨基酸改变 p.Q133X(谷氨酰胺>终止,图2);chr5:60214093区域 c.394_398del,导致氨基酸改变p.L132fs(移码突变,图3)。家系验证结果显示:此双杂合突变分别来自于母亲和父亲,为复合杂合突变;其姐姐存在与其父亲相同的杂合突变。HGMD数据库c.394_398del位点已有报道,c.397C>T位点未见报道,在100名正常人中未发现相关突变。
2 讨论

图2 ERCC8基因c.397C>T突变位点Sanger测序
CS又称侏儒-视网膜萎缩-耳聋综合征,或小头、纹状体小脑钙化和白质营养不良综合征。最主要临床特征是生长发育障碍伴智力障碍、早老化的特殊面容和光敏性皮肤损害等。患者身材呈不匀称性矮小,躯干为主而肢体相对较大,皮下脂肪减少常呈恶病质体型。特殊面容包括皮下脂肪缺如、面部骨骼突出、眼睛深陷、钩状鼻、大耳,类似鸟类的外观[6,7]。神经系统受累表现包括智力障碍或慢性进行性智力减退、小头畸形、共济失调、视听障碍、周围神经病变、不自主运动、痉挛等,周围神经病变一般出现于2岁以后,肌电图检查可显示感觉、运动神经传导速度均减慢[6-8]。眼部病变表现多样,且呈进行性恶化发展,最具特征的是色素性视网膜病变,可有视神经萎缩、白内障、角膜混浊。几乎全部患儿出现感觉神经性耳聋。口腔问题为牙齿发育不全、牙龈炎、龋齿[7]。CS患儿生后1个月即可发生光敏性皮炎,暴露皮肤可发红水肿,后出现色素沉着斑、脱屑。可伴骨骼畸形、心血管和泌尿系统异常,出现脊柱侧弯、高血压、心律失常、动脉粥样硬化、蛋白尿、肾衰等[9,10]。有报道CS患者出现脑动脉硬化和蛛网膜下腔出血,表明脑血管病变可能参与了CS的神经系统损害[10]。CS患者多死于恶病质伴发的疾病或心脑血管疾患[9,10]。本例患儿身材矮小、消瘦,光敏性皮炎,特殊面容,神经系统有精神运动发育迟滞、小头、共济失调、挛缩、肌张力增高,伴随视听障碍、龋齿等,具有CS的典型临床表现。CS患者典型头颅CT显示颅内钙化(最常见于基底节和小脑齿状核)和大、小脑萎缩。头颅MRI表现为脑萎缩和T2WI高信号,最初出现在脑室旁白质、基底节和小脑齿状核,皮层下U形纤维可在病变早期受累,但更多见于病变后期[11]。本例患儿头颅MRI在1岁时正常,5岁复查头颅MRI出现双侧半卵圆中心、侧脑室旁白质对称性脱髓鞘及小脑萎缩改变,可与围产期脑损伤进行鉴别,不符合脑性瘫痪的诊断。本例患儿因家长原因未进行肌电图及眼科和耳科专科检查。对于不明原因精神运动发育迟滞尤其伴有倒退者,建议随访头颅影像情况,以尽早明确诊断,避免误诊。

图3 ERCC8基因c.394_398del突变位点Sanger测序
CS临床诊断要点包括2项主要指标和7项次要指标,主要指标包括:①生长障碍,身高和体质量均落后于同年龄、同性别正常儿童的5个百分位;②进行性神经系统功能异常,神经运动发育迟缓,智力障碍或智力发育落后。次要指标:①侏儒、恶液质体质;②光敏性皮炎,即皮肤对光敏感,接触日光后皮肤红肿继之脱皮,色素沉着,有或无皮肤干燥;③感觉神经性耳聋;④色素性视网膜病或白内障;⑤脱髓鞘性外周神经病;⑥龋齿和/或牙齿脱落;⑦特征性影像学改变,X线检查可见颅骨增厚、骺板硬化、骨盆异常或脊柱侧弯或后凸,头颅CT/MR检查可见大脑白质片状脱髓鞘、小脑萎缩,基底节和小脑周围钙化等。具有2条主要指标加上3条次要指标即可临床诊断CS[12]。本例患儿为7岁学龄期女童,围生期无异常,隐匿发病,主要神经系统表现为精神运动发育迟滞,伴视听障碍、小头、共济失调、挛缩、肌张力增高,头颅MRI显示进行性白质脱髓鞘,尿有机酸和血氨基酸脂酰肉碱谱分析不支持氨基酸、有机酸、脂肪酸代谢障碍,考虑为神经变性病。结合患儿同时有光敏性皮炎、矮小、消瘦、特殊面容、龋齿等表现,考虑为CS,可与其他遗传性白质脑病及Bloom综合征、早老症等鉴别,进一步确诊需要进行基因检测。
CS为DNA损伤后转录修复缺陷导致的疾病,5个不同位点基因CSA(CKN1,ERCC8)、CSB(ERCC6)、XPB、XPD、XPG突变均可以导致CS,这些基因与慢性退行性疾病和氧化应激、衰老相关[13]。也有研究表明线粒体功能与衰老有关,可能参与CS发病[14]。CS具有遗传异质性,临床表型可能差异较大[15]。90%的CS患者是由于CSA和CSB基因缺陷所致[13],ERCC6基因突变约占2/3,ERCC8基因突变占1/3,目前国际已有报道ERCC6突变78种,ERCC8突变30种[6]。国内通过全外显子测序方法,新发现了中国CS患者ERCC6基因突变c.1595A>G (p.Asp532Gly)和 c.1607T>G(p.Leu536Trp),进一步丰富了中国人群的ERCC6基因突变谱[3,5]。相对于Sanger测序,二代高通量测序技术一次可检测数百万条序列,可缩短检测时间、降低测序费用,其发展及广泛应用将使更多的CS患者得到临床及分子遗传学诊断。本例患儿虽具有CS典型的临床表现,但考虑到二代测序的优势,征求患儿家长意见选择了直接进行神经遗传病相关基因的二代测序的方法。
ERCC8基因定位于5q12.1,包含12个外显子,编码的蛋白由396个氨基酸组成。ERCC8为DNA修复因子,参与转录耦合的核苷酸剪接修复,最突出的特点是具有多个WD40重复结构(约含40个氨基酸残基的功能域),该结构在ERCC8发挥生物功能时起到关键作用,不仅是蛋白间相互作用的支架,同时为构建β螺旋结构所必需[16]。目前已报道的致病性突变包括:ERCC8在内的染色体大片段缺失、错义突变、无义突变、移码或截短突变,其中所有错义突变都位于WD40功能域突变种类,但临床表型具有异质性[16-18]。Ting等[17]报道1例CS患儿ERCC8基因同时存在来自于母亲277 kb缺失从而影响整个基因功能和源自父亲外显子4上1,656bp的缺失,此患儿除具有CS表现外,还患有胰岛素依赖性糖尿病。阿拉伯人最常见的ERCC8基因突变类型为Tyr322X,人群携带率达6.79%[19]。Cui等[2]首次在国际上报道1例中国7岁CS男性患儿,临床表现为典型的CS,ERCC8基因检测发现c.551-2A>C 和c.394_398delTTACA两个突变,家系验证分别源自父母。本例患儿临床表现符合典型的CS,ERCC8基因检测亦存在c.394_398del突变,推测此突变有可能是中国CS患儿的常见突变类型,需要更多的样本来证实。本研究发现的c.397C>T突变为首次报道,此突变为终止突变,导致氨基酸改变p.Q133X(谷氨酰胺>终止),对蛋白功能可能影像较大。100例正常人群中检测未发现此突变,不考虑基因多态性。本例患儿的双杂合突变分别来自父母,为复合杂合突变,符合常染色体隐性遗传规律,考虑为致病性突变。本研究扩大了我国CS患者的基因突变谱,对该家庭遗传咨询具有重要指导作用。
CS尚无特殊有效治疗方法,以对症治疗为主。改善营养状况,促进生长和维持机体能量平衡。避免日光曝晒,外出时适当加衣服或防晒霜,保护细胞免受紫外线伤害。在CS小鼠模型中证实存在耳蜗毛细胞受损,对于有条件的CS患儿可进行人工耳蜗植入改善听力[20]。病情进展取决于临床类型,常预后不良。
[1]Hanawalt PC.DNA repair.The bases for Cockayne syndrome[J].Nature,2000,405 (6785): 415-416.
[2]Cui YP,Chen YY,Wang XM,et al.Two novel heterozygous mutations in ERCC8 cause Cockayne syndrome in a Chinese patient [J].Pediatr Neurol,2015,53 (3): 262-265.
[3]Yu S,Chen L,Ye L,et al.Identification of two missense mutations of ERCC6 in three Chinese sisters with Cockayne syndrome by whole exome sequencing [J].PLoS One,2014,9(12): e113914.
[4]谭惠文,吕霞飞,余叶蓉,等.Cockayne综合征一例并文献复习[J].华西医学,2016,31(1): 17-20.
[5]赵振宇,李丽.Cockayne综合征一例[J].中华医学遗传学杂志,2015,32(3): 455-456.
[6]Laugel V.Cockayne syndrome: the expanding clinical and mutational spectrum [J].Mech Ageing Dev,2013,134 (5-6):161-170.
[7]Arenas-Sordo Mde L,Hernández-Zamora E,Montoya-Pérez LA,et al.Cockayne's syndrome: a case report.Literature review [J].Med Oral Patol Oral Cir Bucal,2006,11 (3):E236-238.
[8]Gitiaux C,Blin-Rochemaure N,Hully M,et al.Progressive demyelinating neuropathy correlates with clinical severity in Cockayne syndrome [J].Clin Neurophysiol,2015,126 (7):1435-1439.
[9]Motojima T,Sugita K,Omata T,et al.Clinical examination of renal function in Cockayne syndrome [J].No To Hattatsu,2014,46 (4): 311-314.
[10]Hayashi M,Miwa-Saito N,Tanuma N,et al.Brain vascular changes in Cockayne syndrome [J].Neuropathology,2012,32(2): 113-117.
[11]Simon B,Oommen SP,Shah K,et al.Cockayne syndrome:characteristic neuroimaging features [J].Acta Neurol Belg,2015,115(3): 427-428.
[12]Koob M,Laugel V,Durand M,et al.Neuroimaging in Cockayne syndrome [J].AJNR Am J Neuroradiol,2010,31(9): 1623-1630.
[13]Flores-Alvarado LJ,Ramirez-Garcia SA,Nunez-Reveles NY.The metabolic and molecular bases of Cockayne syndrome [J].Rev Invest Clin,2010,62(5): 480-490.
[14]Scheibye-Knudsen M ,Croteau DL,Bohr VA.Mitochondrial deficiency in Cockayne syndrome [J].Mech Ageing Dev,2013,134(5-6): 275-283.
[15]Laugel V,Dalloz C,Durand M,et al.Mutation update for theCSB/ERCC6andCSA/ERCC8genes involved in Cockayne syndrome [J].Hum Mutat,2010,31(2): 113-126.
[16]Koch S,Garcia GO,Assfalg R,et al.Cockayne syndrome protein A is a transcription factor of RNA polymeraseⅠand stimulates ribosomal biogenesis and growth [J].Cell Cycle,2014,13 (13): 2019-2037.
[17]Ting TW,Brett MS,Tan ES,et al.Cockayne syndrome due to a maternally-inherited whole gene deletion ofERCC8and a paternally-inheritedERCC8exon 4 deletion [J].Gene,2015,572 (2): 274-278.
[18]Conchello-Monleon R,Pena-Segura JL,Tello-MartinA,et al.Cockayne syndrome: a new mutation in theERCC8gene [J].Rev Neurol,2012,55 (4): 250-251.
[19]Khayat M,Hardouf H,Zlotogora J,et al.High carriers frequency of an apparently ancient founder mutation p.Tyr322X in theERCC8gene responsible for Cockayne syndrome among Christian Arabs in Northern Israel [J].Am J Med Genet A,2010,152A (12): 3091-3094.
[20]Nagtegaal AP,Rainey RN,van der Pluijm I,et al.Cockayne syndrome group B (Csb) and group a (Csa) deficiencies predispose to hearing loss and cochlear hair cell degeneration in mice [J].J Neurosci,2015,35 (10): 4280-4286.
2017-03-28)
(本文编辑: 梁 华)
A case of Cockayne syndrome caused byERCC8gene mutation
MA Xiuwei1,ZHAO Jiayan2,GU Ruijie1,FENG Zhichun1(1.Department of Neurology and Development,Bayi Children’s Hospital Affiliated to PLA Army General Hospital,Beijing 100700,China;2.Department of Pediatrics,Third Af fi liated Hospital of Xinxiang Medical College,Xinxiang 453000,Heman,China)
ObjectiveTo explore the clinical,radiological and gene mutation features ofERCC8gene in one patient with Cockayne syndrome.MethodsClinical and radiological data of a girl diagnosed with Cockayne syndrome through gene detection were retrospectively analyzed.Next-generation sequencing was used to detect genetic cause.Sanger sequencing was used to con fi rm the candidate variants and detect mutations in her parents and sister.ResultsThe patient showed psychomotor retardation,growth failure,special face,and light sensitivity.Neurological examination revealed noticeable developmental delay,motor impairment,spastic paralysis,and cerebellar ataxia.Brain MRI revealed symmetrical demyelination of bilateral centrum semiovale and periventricular white matter.The cerebellum was atrophic.The patient was found to have compound heterozygous mutations of c.397C>T(p.Q133X) and c.394_398del(p.L132fs).Sanger sequencing showed these two mutations were inherited from her mother and father respectively.ConclusionsNext-generation sequencing technology is a useful tool for the detection of mutation inERCC8gene,which is valuable for the diagnosis of Cockayne syndrome.These two mutations expanded the mutation spectrum of Cockayne syndrome in Chinese population.
Cockayne syndrome; clinical feature;ERCC8gene; gene mutation
10.3969/j.issn.1000-3606.2017.11.004
