石墨烯在Al2O3(0001)表面生长的模拟研究∗
2017-11-10李锦锦李多生洪跃邹伟何俊杰
李锦锦 李多生 洪跃 邹伟 何俊杰
(南昌航空大学材料科学与工程学院,南昌 330063)
石墨烯在Al2O3(0001)表面生长的模拟研究∗
李锦锦 李多生†洪跃 邹伟 何俊杰
(南昌航空大学材料科学与工程学院,南昌 330063)
(2017年4月23日收到;2017年7月25日收到修改稿)
CH4,蓝宝石(α-Al2O3)(0001),吸附分解,密度泛函理论
1 引 言
蓝宝石室温下具有10 eV的带隙,是一种综合性能优异的绝缘材料,具有高硬度、高熔点、高透波性、化学性能稳定等特点.蓝宝石晶体在红外军用装置、导弹、潜艇、卫星空间技术、探测和高功率强激光等优良窗口材料方面具有较为广泛的应用[1−3].石墨烯具有极好的导电性、导热性、强度及透波性等优异性能,将其作为薄膜材料与蓝宝石结合在一起,在高马赫飞行器的红外窗口材料、传感器[4−6]、半导体器件、透明电极等方面[7,8],都有较大的应用前景.目前高质量石墨烯的生长多采用化学气相沉积法,通过甲烷在衬底表面的吸附分解,最终形成石墨烯[9,10].甲烷在金属衬底表面上生长制备高质量石墨烯的研究已经很多,主要是利用金属对甲烷的催化分解作用或利用金属对C原子的固溶析出原理[11−13].在非金属衬底表面尤其是在蓝宝石衬底上生长制备石墨烯薄膜还比较困难,对此还没有进行过相关的模拟研究.要在蓝宝石衬底表面生长制备石墨烯,首先要考虑甲烷在蓝宝石衬底表面上的吸附分解过程,以及石墨烯在衬底表面的形核方式.我们采用Materials Studio软件对上述过程进行第一性原理计算和分子动力学(MD)模拟.
2 理论模型与计算方法
计算时采用Materials Studio软件的CASTEP模块[14,15].CASTEP模块是基于密度泛函理论,采用总能量赝势法和平面波展开法进行计算[16,17].交换与关联能采用广义梯度近似法下的Perdew-Burke-Ernzerhof[18]泛函来描述.为了使计算有较好的收敛结果,采用Broyden-Fletcher-Goldfarb-Shanno[19]优化算法进行结构优化.经过收敛性测试后,取收敛标准:平面波展开的截断能取400 eV,布里渊区网格化密度取3×3×1.原子结构优化中作用于原子上的受力小于0.1 eV·nm−1,单原子能量小于5.0×10−6eV/atom,原子位移变化小于5.0×10−5nm;自洽场迭代过程收敛精度为1.0×10−6eV/atom.计算CH4分解的过渡态时采用CASTEP模块里的COMPLETE LST/QST程序[20].所有计算在0 K下进行.CH4及其分解产物CH3,CH2,CH基团和C,H原子在α-Al2O3(0001)表面的吸附能(Eads)通过(1)式进行计算.
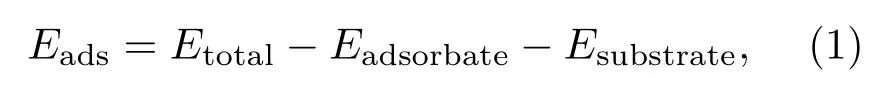
(1)式中,Eads表示CH4分子、CH3、CH2、CH基团或C、H原子在α-Al2O3(0001)表面的吸附能,Etotal表示吸附后的体系总能量,Eadsorbate表示CH4分子、CH3、CH2、CH基团或C、H原子的能量,Esubstrate表示α-Al2O3(0001)表面衬底模型的能量.
同时对α-Al2O3(0001)表面上石墨烯形核初期的活跃基团做了MD模拟.MD模拟是基于CASTEP里的Dynamics程序,采用NVT系统,在1300 K下运行2.0 ps,步长1.0 fs,温度在经历0.01 ps后达到平衡.
3 计算结果及分析
3.1 α-Al2O3表面模型
对α-Al2O3的原始晶胞进行结构优化,得到优化后的晶胞参数a=b=4.807 Å,c=13.122 Å,与标样测得的参数a=b=4.766 Å,c=12.996 Å基本一致[21].鉴于α-Al2O3(0001)表面是其最稳定的非极性面且原子结构简单[22,23],以此面构建(2×2)的超晶胞结构作为蓝宝石的表面模型,如图1(a)所示:模型包含3层O原子和6层Al原子,共计60个原子.其中模型下方的4层非表面层原子固定,模型上方5层表面层原子位置可变[22].同时建立12 Å的真空层,以避免晶层间原子之间的相互影响.
图1(a)和图1(b)分别是α-Al2O3(0001)表面结构优化前后的对比图.表1是结构优化后原子层层间距变化的具体数据.结合图1和表1,可以看出经过结构优化后α-Al2O3(0001)表面层原子向内收敛.从表1可以看出,最外层的4层原子层厚度有较大的弛豫,尤其是第一层原子层厚度变化较大.随着向内部的深入,层间距变化值不断减小,这与前人的理论计算相符合[24−26].然而这些理论计算值的第一层层间距变化在−86%—−93.8%之间与实验值−51%[27]存在差距,这是由于实验值测量时环境中含有的氢或者羟基造成的,在文献[28]有较好的解释.因此我们建立的衬底模型是合理的.
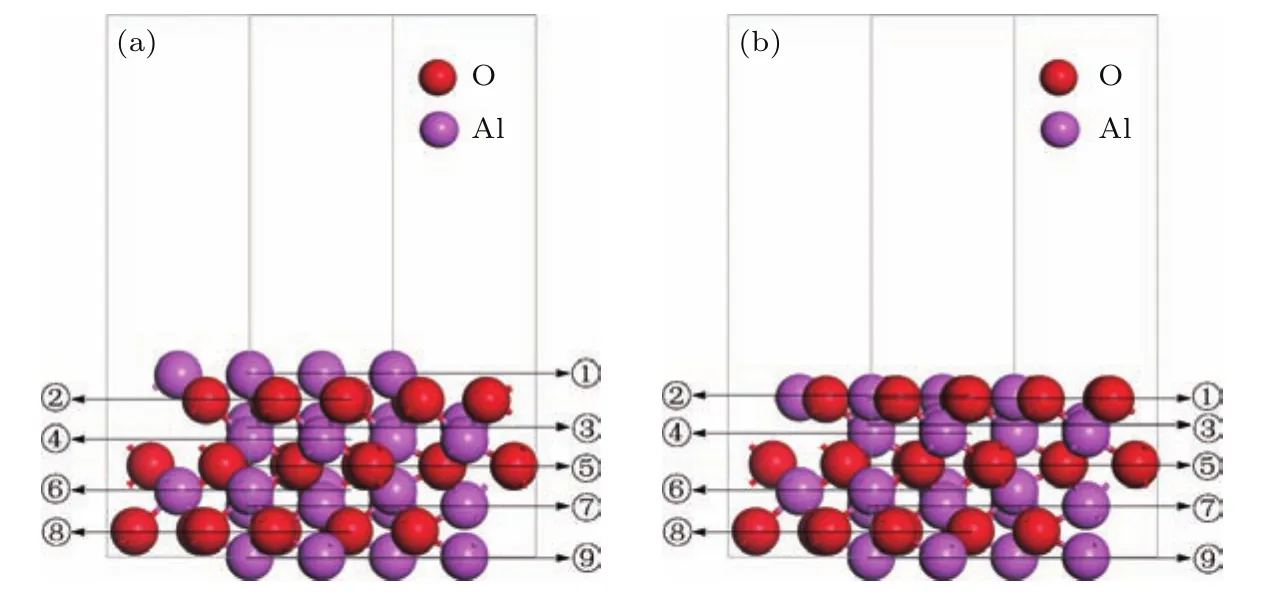
图1 (网刊彩色)α-Al2O3(0001)表面模型 (a)结构优化前;(b)结构优化后Fig.1.(color online)The surface model of α-Al2O3(0001):(a)Before structural optimization;(b)after structural optimization.
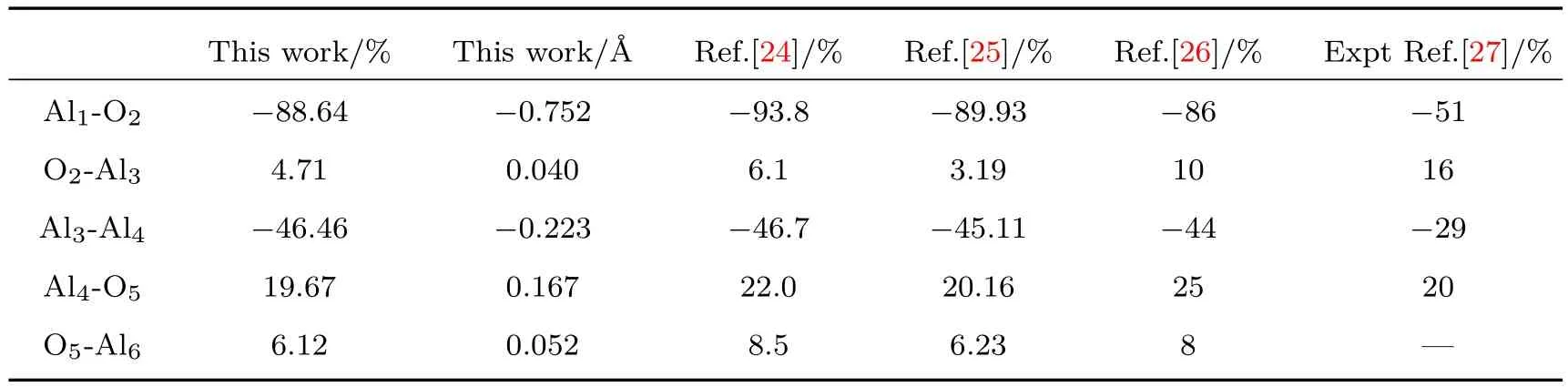
表1 α-Al2O3(0001)表面结构优化后层间距变化(幅度百分比 =(优化后的距离−优化前的距离)/优化前的距离×100%)Table 1.The interlayer relaxations of the Al2O3(0001)surface as a function of slab thickness(magnitude percentage=(relaxed distances−unrelaxed distances)/unrelaxed distances×100%).
3.2 CH4分解产物的稳定吸附位
CH4在α-Al2O3(0001)表面分解后形成的基团及原子的最稳定吸附位和吸附构型如图2所示,表2列出了它们的吸附能.CH4在α-Al2O3(0001)表面上的分解主要由以下4步完成:CH4→CH3+H,CH3→CH2+H,CH2→CH+H,CH→C+H.甲烷在衬底表面的分解会产生过渡基团和原子,分别为CH3,CH2,CH基团和C,H原子.分别计算这些基团及原子在衬底表面原子的顶位、桥位、穴位下稳定体系的能量,取体系能量最低的结构作为最稳定吸附结构,此时这些基团及原子与衬底表面相互作用最强.其中CH3基团最稳定吸附位在表面O原子的顶位,吸附能为−2.428 eV.CH2,CH基团最稳定吸附位在表面O,Al原子之间的桥位,吸附能分别为−4.460和−3.940 eV.上述基团稳定吸附后,C—H键键长增加,削弱了C—H键的键能,有利于基团中C—H键进一步断裂分解.C,H原子最稳定吸附位在表面O原子的顶位,吸附能分别为−4.903和−4.083 eV.基于这些数据,下面进一步计算了CH4在α-Al2O3(0001)表面的分解能.

表2 各基团及原子的吸附能及最稳定吸附位Table 2.The adsorption energies and stable site of groups and atoms.
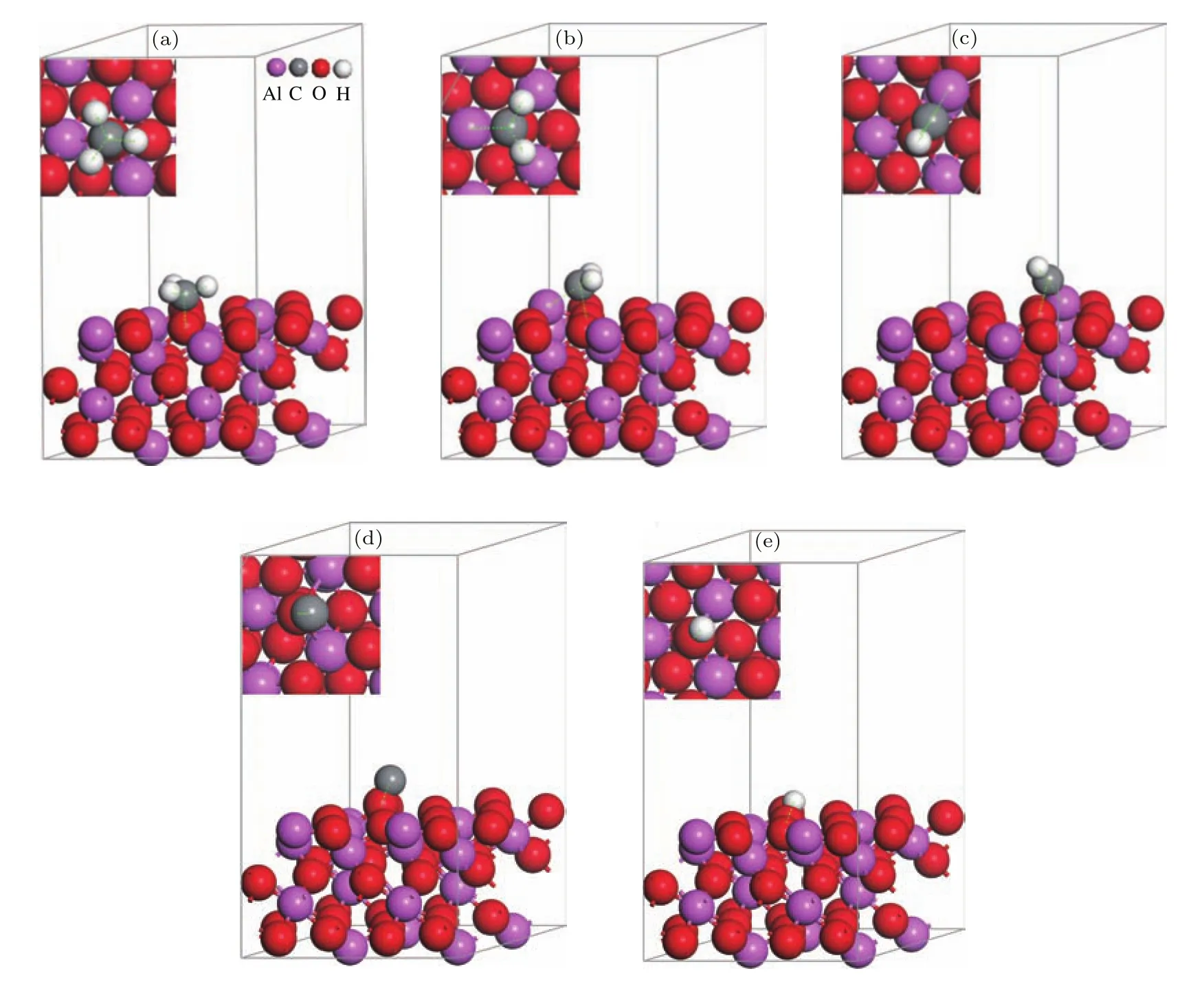
图2 (网刊彩色)各基团及原子在α-Al2O3(0001)表面稳定吸附位和吸附构型示意图(顶视图和侧视图) (a)CH3的稳定吸附位;(b)CH2的稳定吸附位;(c)CH的稳定吸附位;(d)C的稳定吸附位;(e)H的稳定吸附位Fig.2.(color online)The schematic of different groups and atoms on the surface of α-Al2O3(0001)based on the most stable site(shown looking down onto the surface and sideways):(a)The stable adsorption site of CH3;(b)the stable adsorption site of CH2;(c)the stable adsorption site of CH;(d)the stable adsorption site of C;(e)the stable adsorption site of H.
3.3 CH4在α-Al2O3(0001)表面的分解能
CH4在α-Al2O3(0001)表面分解的每步反应热和能垒如表3所列.图3是反应中各过渡态能量变化示意图.研究发现,CH4的逐步分解过程中,首先由CH4分解出一个H原子后吸附在O原子的顶位,剩下的CH3基团吸附在临近O原子的顶位,为吸热过程,该过程反应热为2.873 eV,活化能为3.348 eV;接着CH3基团中的一个等位C—H键断裂分解出一个H原子后成CH2基团,吸附在表面的O与Al原子之间的桥位,分解出的H原子吸附在CH2基团附近的O原子的顶位,为吸热过程,该步反应热为2.777 eV,活化能为2.787 eV;然后CH2分解出一个H原子后成CH基团吸附在O与Al原子之间的桥位,H原子吸附在CH基团附近的O原子的顶位,为吸热过程,该步反应热为1.729 eV,活化能为1.796 eV;最后CH基团分解出一个H原子后以单原子C吸附在O原子的顶位,H原子吸附在C原子附近的O的顶位,为吸热过程,该步反应热为1.659 eV,活化能为3.985 eV.在这4步反应中,第三步反应所需的反应能量和反应活化能垒最低,第四步反应所需的反应能量和反应活化能垒最高.
石墨烯在α-Al2O3(0001)表面的形核,通常认为是以CH4在衬底表面分解出的C为基本单元.C在衬底表面迁移聚集并偶联到一定尺度后,石墨烯形核并逐渐生长[29−32].通过对图3和表3的分析发现,CH4在α-Al2O3(0001)表面的分解是吸热过程.随着CH4在α-Al2O3(0001)表面脱氢的进行,整个体系能量在不断升高.CH4彻底分解的终态比初态能量升高了9.073 eV,在热力学上,这个过程是很难发生的.且每一步的脱氢反应能垒都在1.7 eV以上,在由CH→C的脱氢过程中,能垒高达3.9 eV,这就需要较高的能量起伏才能促使CH基团跃过能垒分解出C和H.上述模拟数据研究表明,CH4在α-Al2O3(0001)表面彻底分解出C较困难,这样在α-Al2O3(0001)表面很难提供足够的C用于石墨烯形核,石墨烯在α-Al2O3(0001)表面的形核活跃因子不能是C.
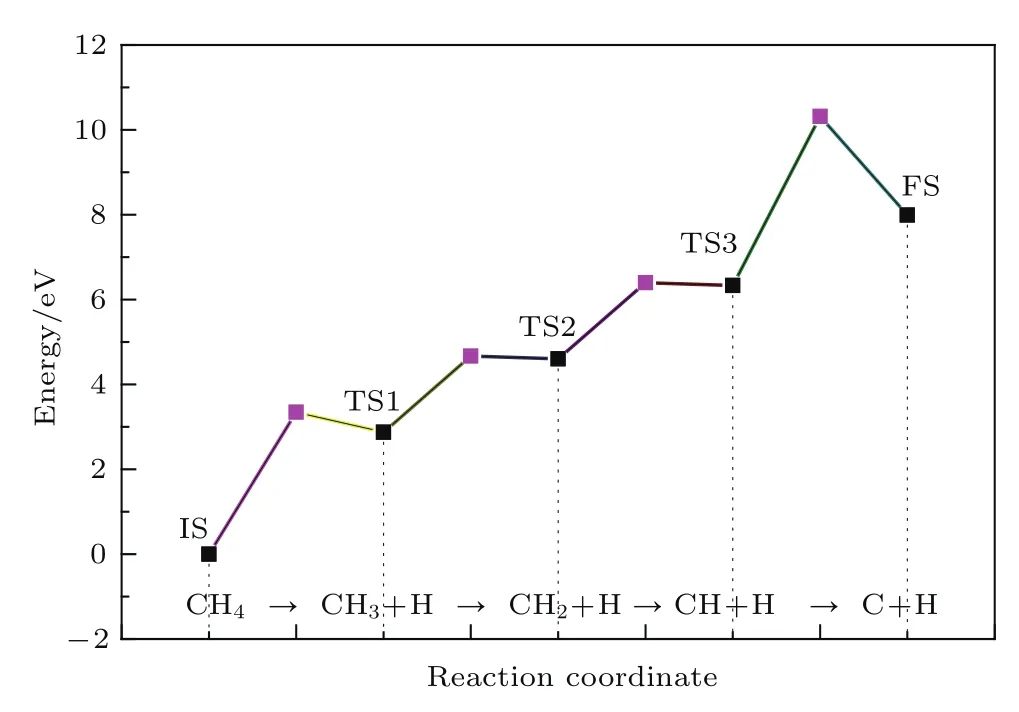
图3 (网刊彩色)CH4在α-Al2O3(0001)表面分解能Fig.3.(color online)Decomposition Energy of CH4 on α-Al2O3(0001)surface.
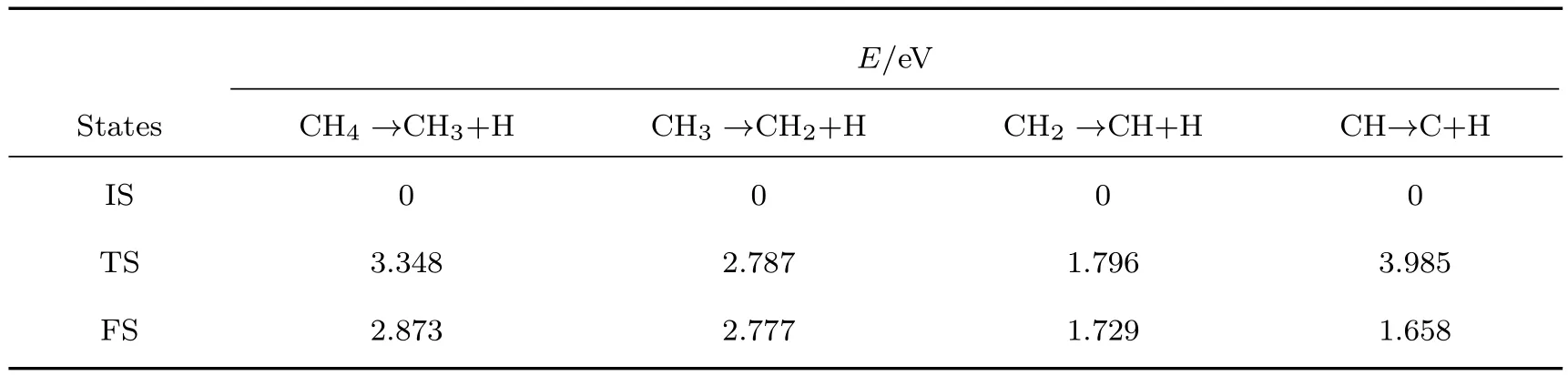
表3 CH4分解的初始态、过渡态、终止态相对能(E)Table 3.Relative energies(E)of the initial state(IS),transition state(TS),and fi nal state(FS)for CH4 decomposition.
3.4 石墨烯形核模式
用化学气相沉积法生长石墨烯的制备过程中,通常将C原子在衬底表面的弛豫迁移偶联作为石墨烯形核的起点[29−32].这主要利用了CH4在一些衬底上的分解是放热过程或者本身衬底表面对CH4分解出的C的溶解度较高.如在Pd和Ru活泼金属衬底表面上,CH4的分解是放热过程,即随着CH4脱氢的进行反应体系的能量不断降低[33,34],衬底表面上利于C的存在,能为石墨烯形核提供足够的碳源.因此在这些金属上石墨烯的形核是以CH4裂解出的C为活跃因子,通过C在衬底表面不断聚集偶联生长最终形成石墨烯.在Ni表面上,CH4的分解与在α-Al2O3(0001)表面上的分解同样是吸热过程,不利于衬底表面C的生成,但Ni对C有着较高的溶解度,利于分解出的C溶解于衬底表面,通过溶解析出过程使石墨烯在Ni表面形核[35,36].首先对于CH4在α-Al2O3(0001)表面上的分解是吸热过程,其次α-Al2O3衬底对C的溶解度很低,这些因素均不利于C在α-Al2O3(0001)表面上的存在,因此石墨烯在α-Al2O3(0001)表面上形核模式不同于上述的方式.
通过计算发现,在α-Al2O3(0001)表面上,由CH偶联成C2H2的能垒只有1.822 eV,且该过程是放热过程,偶联后体系能量降低4.283 eV.进一步计算了由C2H2偶联一个CH转变成C3H3的过程,该过程能垒2.689 eV,体系能量降低3.740 eV,数据如表4所列.在α-Al2O3(0001)表面上,CH基团之间相互偶联成(CH)x结构,不仅能垒小于CH→C的分解过程,同时体系能量降低,该过程更易进行.我们认为在α-Al2O3(0001)表面上,石墨烯形核的初期是围绕着CH基团进行的而不是C,在Ru(0001)面得到类似的实验研究[37].在最终的石墨烯形成之前,α-Al2O3(0001)表面上的CH基团之间首先相互偶联在一起形成能量较低的(CH)x结构.当然,最终石墨烯的形成还是要经过(CH)x结构的脱H过程.只是脱H的过程是在一定尺寸的(CH)x结构形成之后进行的,类似的研究也反映了该过程的存在的可能[38].
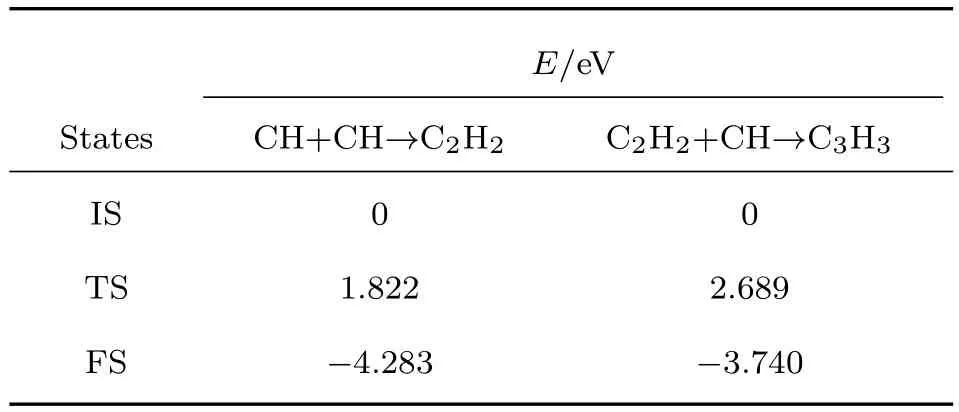
表4 (CH)x初始态、过渡态、终止态的相对能(E)Table 4.Relative E of the IS,TS,and FS for(CH)x.
3.5 CH作为石墨烯形核活跃因子的MD模拟
为了进一步验证石墨烯形核的初期是围绕着CH基团进行的而不是C,进一步开展了MD模拟.在α-Al2O3(0001)表面上覆盖CH基团,其覆盖度为0.41 ML,采用NVT系统在1300 K的温度下运行2.0 ps时长.模拟结果如图4所示,可以看出在α-Al2O3(0001)表面上CH基团的分布出现了明显的特点:1)高温下α-Al2O3(0001)表面上的CH基团并没有发生C—H键的断裂,α-Al2O3(0001)表面没有出现单独存在的C原子,虽然CH→C过程分解的能垒为3.985 eV,此能垒足以在1300 K的温度下克服,但裂解后的CH也有很大的概率重新结合在一起,这样,并没有观察到CH完整的分解过程;2)在α-Al2O3(0001)表面上,出现了较多的C2H2结构和少量C3H3结构,如图4中黑色圆圈中所标注,这表明CH基团很容易结合在一起存在于α-Al2O3(0001)表面,CH基团通过结合形成(CH)x结构后,降低了衬底上的界面能,整个体系能量降低,有利于CH在衬底表面的存在.因此衬底表面大量(CH)x结构的存在是石墨烯形核的基础,CH是石墨烯形核的活跃因子.
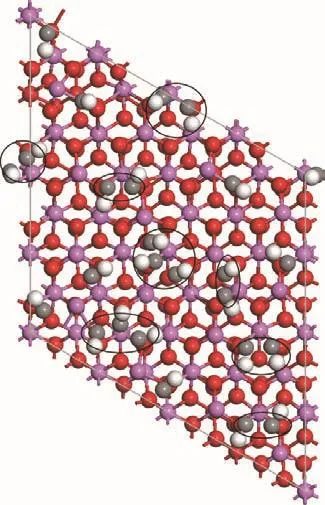
图4 (网刊彩色)覆盖CH的α-Al2O3(0001)表面MD模拟结果Fig.4.(color online)MD simulation result of α-Al2O3(0001)surface covered with CH.
4 结 论
建立蓝宝石α-Al2O3(0001)表面模型,采用密度泛函理论对CH4在其表面上的吸附分解以及石墨烯形核机理进行研究.发现CH3基团及C,H原子优先吸附在O原子的顶位,吸附能分别为−2.428,−4.903,−4.083 eV.CH2,CH基团优先吸附在O,Al原子之间的桥位,吸附能分别为−4.460,−3.940 eV.CH4在α-Al2O3(0001)表面的分解是吸热过程,且由CH→C过程能垒较高,这些因素都不利于C在α-Al2O3(0001)表面的存在.在α-Al2O3(0001)表面上大量存在的CH基团是石墨烯形核的活跃基团,这些基团在衬底表面优先形成能量较低的(CH)x结构,形成石墨烯形核前驱体.
[1]Tang S H,Cao X X,He L,Zhu W J 2016Acta Phys.Sin.65 146201(in Chinese)[唐士惠,操秀霞,何林,祝文军2016物理学报65 146201]
[2]Stojchevska L,Vaskivskyi I,Mertelj T,Kusar P,Svetin D,Brazovskii S 2014Science344 177
[3]You F,Ji L,Xie Q L,Wang Z,Yue H W,Zhao X J,Fang L,Yan S L 2010Acta Phys.Sin.59 5035(in Chinese)[游峰,季鲁,谢清连,王争,岳宏卫,赵新杰,方兰,阎少林2010物理学报59 5035]
[4]Wang X,Zhi L G,Mullen K 2008Nano Lett.8 323
[5]Simon P,Gogotsi Y 2008Nat.Mater.7 845
[6]Kim K S,Zhao Y,Jang H,Lee S Y,Kim J M,Kim K S,Ahn J,Kim P,Choi J Y,Hong B H 2009Nature457 706
[7]Zheng H W 2006Ph.D.Dissertation(Hefei:University of Science and Technology of China)(in Chinese)[郑海务2006博士毕业论文(合肥:中国科学技术大学)]
[8]Zhang Y B,Tan Y W,Stormer H L,Kim P 2005Nature438 201
[9]Li X S,Cai W W,An J H,Kim S,Nah J,Yang D X,Piner R,Velamakanni A,Jung I,Tutuc E,Banerjee S K,Colombo L,Ruo ffR S 2009Science324 1312
[10]Obraztsov A N 2009Nat.Nanotech.4 212
[11]Hofmann S,Csanyi G,Ferrari A C,Payne M C,Robertson J 2005Phys.Rev.Lett.95 036101
[12]Bhaviripudi S,Jia X T,Dresselhaus M S,Kong J 2010Nano Lett.10 4128
[13]Riikonen S,Krasheninnikov A V,Halonen L,Nieminen R M 2012J.Phys.Chem.C.116 5802
[14]Smith J R,Hong T,Smith J R,Srolovitz D J 1995Acta Metall.Mater.43 2721
[15]Jiang Z,Pan Q,Li M,Yan T,Fang T 2014Appl.Surf.Sci.292 494
[16]Blochl P E 1994Phys.Rev.B50 17953
[17]Kresse G,Joubert D 1999Phys.Rev.B59 1758
[18]Perdew J P,Burke K,Ernzerhof M 1996Phys.Rev.Lett.77 3865
[19]Fischer T H,Almlof J 1992J.Phys.Chem.B96 9768
[20]Halgren T A,Lipscomb W N 1977Chem.Phys.Lett.49 225
[21]Finger L W,Hazen R M 1978Appl.Phys.49 5823
[22]Baxter R,Reinhardt P,López N,Illas F 2000Surf.Sci.445 448
[23]Wander A,Searle B,Harrison N M 2000Surf.Sci.458 25
[24]Carrasco J,Gomes J R B,Illas F 2004Phys.Rev.B69 064116
[25]Rohmann C,Metson J B,Idriss H 2011Surf.Sci.605 1694
[26]Chiang H N,Nachimuthu S,Cheng Y C,Damayanti N P,Jiang N P 2016Surf.Sci.363 636
[27]Guenard P,Renaud G,Barbier A,Gautiersoyer M 1998Surf.Rev.Lett.5 321
[28]Hass K C,Schneider W F,Curioni A,Andreoni W 1998Science282 265
[29]Li X,Cai W,Colombo L,Ruo ffR S 2009Nano Lett.9 4268
[30]Wu P,Zhang W,Li Z,Yang J,Hou J G 2010J.Chem.Phys.133 071101
[31]Chen H,Zhu W,Zhang Z 2010Phys.Rev.Lett.104 186101
[32]Niu T,Zhou M,Zhang J,Feng Y,Chen W 2013J.Am.Chem.Soc.135 8409
[33]Zhang C J,Hu P 2002J.Phys.Chem.116 322
[34]Ciobîca I M,Frechard F,van Santen R A,Kleyn A W,Hafner J 2000J.Phys.Chem.B104 3364
[35]Watwe R M,Bengaard H S,Rostrup-Nielsen J R,Dumesic J A,Nørskov J K 2000J.Catal.189 16
[36]Xu J,Saeys M 2008J.Phys.Chem.C112 9679
[37]Gao M,Zhang Y F,Huang L,Pan Y,Wang Y,Ding F,Lin Y,Du S X,Gao H J 2016Appl.Phys.Lett.109 131604
[38]Treier M,Pignedoli C A,Laino T,Rieger R,Mullen K,Passerone D 2010Nat.Chem.3 61
PACS:71.15.Mb,68.43.–h,63.20.dk,73.20.Hb DOI:10.7498/aps.66.217101
*Project supported by the National Natural Science Foundation of China(Grant No.51562027)and the Jiangsu Key Laboratory of Precision and Micro Manufacturing Technology Foundation,China(Grant No.JKL2015001).
†Corresponding author.E-mail:duosheng.li@nchu.edu.cn
Growth of graphene on Al2O3(0001)surface∗
Li Jin-Jin Li Duo-Sheng†Hong Yue Zou WeiHe Jun-Jie
(School of Materials Science and Engineering,Nanchang Hangkong University,Nanchang 330063,China)
d 23 April 2017;revised manuscript
25 July 2017)
At present,high quality graphene is synthesized mainly by chemical vapor deposition.It is crucial to decompose and adsorb methane(CH4)on the surface of substrate before CH4grows into graphene.The graphene is grown mainly on metal substrate due to the catalytic effect of metal.It is difficult to grow graphene thin film on the surface of non-metallic substrate,especially on the surface ofα-Al2O3(0001).In this paper,the density functional theory based generalized gradient approximation method is applied to simulating the nucleation of graphene onα-Al2O3(0001)surface,synthesized by chemical vapor deposition.First,we establish a scientificα-Al2O3(0001)surface model,then simulate the decomposition process of CH4onα-Al2O3(0001)surface by calculating the adsorption sites and adsorption configurations of groups and atoms.Finally,we investigate the groups of CH4decomposition and atom coupling process onα-Al2O3(0001)surface.The results show that the CH3groups,C and H atoms are preferentially adsorbed at the top of the O atoms,and the adsorption energies are−2.428 eV,−4.903 eV,and−4.083 eV,respectively.The CH2and CH groups are preferentially adsorbed on the bridge between O and Al atoms with the adsorption energies of−4.460 eV and−3.940 eV,respectively.The decomposition of CH4onα-Al2O3(0001)surface is an endothermic process.It requires higher energy and cross reactive energy barrier for CH4to be completely decomposed into C atom,which makes it difficult that the C atom stays on the substrate surface.The coupling process among CH groups on the surface ofα-Al2O3(0001)is an exothermic process.When CH and CH groups are coupled,the energy of the system decreases by 4.283 eV.When(CH)2and CH groups are coupled,the energy of the system decreases by 3.740 eV.The(CH)xcan be obtained by continuous migration and coupling between the CH groups on the surface of theα-Al2O3(0001),and(CH)xgroup is a precursor of graphene growth.The energy of the system decreases in the process.The above results show that the activated atom or group of graphene nucleation is not C atom but CH group.The CH group migration and aggregation on the surface ofα-Al2O3(0001)give priority to the formation of lower energy(CH)xstructure.In order to better understand the microscopic growth process of graphene on sapphire,it is important to study the role of(CH)xin the surface of sapphire for revealing the nucleation mechanism of graphene.
CH4,sapphire(α-Al2O3)(0001),adsorption and decomposition,density functional theory
基于密度泛函理论的广义梯度近似法,对用化学气相沉积法在蓝宝石(α-Al2O3)(0001)表面上生长石墨烯进行理论研究.研究结果表明:CH4在α-Al2O3(0001)表面上的分解是吸热过程,由CH4完全分解出C需要较高能量及反应能垒,这些因素不利于C在衬底表面的存在.在α-Al2O3(0001)表面,石墨烯形核的活跃因子并不是通常认为的C原子,而是CH基团.通过CH基团在α-Al2O3(0001)表面上的迁移聚集首先形成能量较低的(CH)x结构.模拟研究(CH)x对揭示后续石墨烯的形核生长机理具有重要意义.
10.7498/aps.66.217101
∗国家自然科学基金(批准号:51562027)和江苏省精密与微细制造技术重点实验室基金(批准号:JKL2015001)资助的课题.
†通信作者.E-mail:duosheng.li@nchu.edu.cn
©2017中国物理学会Chinese Physical Society
