利用Red/ET同源重组构建新型CCR5Δ32打靶载体
2017-11-06卢智勇杨卓顺丁妍袁雅红王小莉于莉李东升
卢智勇,杨卓顺,丁妍,袁雅红,王小莉,于莉,李东升
湖北省十堰市太和医院(湖北医药学院附属医院)生命科学研究所,湖北 十堰 442000
利用Red/ET同源重组构建新型CCR5Δ32打靶载体
卢智勇,杨卓顺,丁妍,袁雅红,王小莉,于莉,李东升
湖北省十堰市太和医院(湖北医药学院附属医院)生命科学研究所,湖北 十堰 442000
目的:目前应用TALENs或CRISPR-Cas基因编辑技术修饰CCR5基因以获得抗HIV功能的研究策略基本是破坏CCR5基因而不是制造CCR5Δ32这样天然存在的抗HIV基因型。为此,我们尝试通过细菌内同源重组技术构建一个新颖的CCR5Δ32的打靶载体,以便能够用于制备CCR5Δ32基因突变细胞系。方法:首先利用Red/ET同源重组系统将rpsl-neo片段插入细菌人工染色体(BAC)的CCR5基因中,随后再次利用同源重组将一个不含32bp区域的非选择性52bp片段替换该rpsl-neo片段,从而将BAC中的CCR5基因改造成CCR5Δ32基因型,最后将loxp-neo-loxp抗性基因插入CCR5基因的第2内含子区域,制备出含有抗性基因的CCR5Δ32打靶载体,该载体可在Cre酶作用下删除neo基因但只留下一个loxp在基因的内含子区。结果:rpsl-neo片段成功插入CCR5之Δ32区并为非选择性52bp片段替换,loxp-neo-loxp成功插入内含子区。结论:通过Red/ET同源重组技术获得CCR5Δ32-loxp-neo-loxp打靶载体。
细菌人工染色体;CCR5;同源重组;重组工程
尽管针对人类免疫缺陷病毒(human immuno⁃deficiency virus,HIV)的预防和治疗到目前为止已取得一定进展,如抗病毒药物和疫苗,但成本高昂疗效有限,因此很有必要深入研究新型HIV治疗靶标和治疗措施。CCR5(C-C chemokine re⁃ceptor type 5)是HIV-1在靶细胞上的关键受体之一,可以介导HIV对靶细胞的感染[1]。在白人种族中存在CCR5的一种突变体即CCR5Δ32,与野生型CCR5相比缺失了32个碱基,从而使得该类人群天然对HIV具有抵抗力[2]。自从发现这个现象后,以CCR5为靶点的研究层出不穷,如CCR5小分子拮抗剂可以阻断HIV-1的感染并开展相应的临床试验,另外通过抗体、核酶及siRNA敲低CCR5表达水平也可以对抗HIV-1的感染[3-5]。近来随着ZFNs、TALENs和CRISPR-Cas9基因编辑技术的飞速发展,针对人类胚胎干细胞(human embryonic stem cells,hESCs)和T细胞进行CCR5的敲除亦屡见报道[6-8]。这3类基因编辑技术都是利用合成的核酸内切酶在指定的DNA靶位切割双链后造成双链缺口即DSB,然后利用细胞本身的修复机制引入突变包括随机敲除和定点插入,从而实现对细胞基因组的改造。通常,随机敲除发生的概率远较定点删除和插入为高,因此绝大多数学者都是针对目的细胞如hESCs、造血干细胞、T细胞进行CCR5基因的随机敲除突变[6-8],仅有1篇报道联合运用piggyBAC技术将CCR5基因改造为CCR5Δ32这一天然存在的突变形式[9]。尽管目前认为敲除CCR5基因不会对机体产生明显的有害作用,但是如果能够像CCR5Δ32既保留类似CCR5的功能又可获得HIV抗感染能力,将是非常完美的研究方向和治疗靶点。但是由于定点删除的难度很高,很少有学者进行这种基因修饰,因此有必要构建一种新型载体联合基因编辑技术来提高靶细胞产生CCR5Δ32基因型的发生率。为此,我们通过联合运用细菌人工染色体(bacterial artificial chromosome,BAC)和细菌内同源重组技术,将BAC上的野生型CCR5基因改造成含有抗性基因的CCR5Δ32基因型。
细菌内同源重组是一项高效特异地利用大肠杆菌工程菌构建载体的分子克隆技术[10-11],由于采用的是细菌基因组自身表达的同源重组酶进行质粒或BAC的修饰改造,因此它不需要任何限制性内切酶及其位点[12-13],这个优势使得它可以用来构建比较复杂或没有合适内切酶位点的载体。而且,考虑到BAC的巨大容量可以包含整个CCR5基因的外显子、内含子,以及如启动子等基因调控单元,因此在BAC基础上构建的某个特定基因的载体,由于包含基因调控单元,更类似于内源性的基因序列,对于基因打靶或基因编辑,相对普通分子克隆技术构建的打靶载体而言具有更大优势(比如包含内源性调控元件、更长的同源臂)。BAC是一种以F质粒为基础建构而成的细菌染色体克隆载体,常用来克隆150kb左右的DNA片段,最多可保存300kb碱基。目前主要用于大片段基因组文库的构建和大的基因簇的相关研究,并在各类生物基因组计划中发挥重要作用。细菌内同源重组技术是将来源于噬菌体的3个与重组有关的基因整合进大肠杆菌染色体组中获得的。简言之,3个基因中的gam基因负责保护外源导入的DNA片段不被细菌自身的RecBCD核酸酶水解;而exo基因具备核酸外切酶活性,负责将双链切割出一个单链突出末端;随后beta基因就与该突出末端结合,将具有同源臂的外源DNA片段与靶位点DNA进行同源重组。传统的载体构建技术需要PCR扩增、酶切、连接等程序和相应的酶,尤其是需要相应的酶切位点,受限严重,而细菌内同源重组技术则完全避免了酶切位点的限制,且高效精确,因此非常适合用于基因改造的打靶载体构建[13-14]。
本研究中,我们采用类似Zhang等报道的重组技术[15-17],首先针对CCR5基因中的Δ32区域设计并合成具备同源臂的rpsl-neo片段,将其电转入含有BAC和pRedET的大肠杆菌DH10B中,通过该菌内的同源重组系统将rpsl-neo片段插入BAC的CCR5中并替换掉含有Δ32的52bp区域(图1,step1);随后设计并合成一段也具有同源臂的不含Δ32的180bp小片段(与野生型CCR5的Δ32区除没有32bp外完全相同),同样利用同源重组系统以180bp小片段替换掉rpsl-neo,从而获得CCR5Δ32基因型(图1,step2);最后利用同样的技术将loxp-neo-loxp抗性基因插入CCR5基因的第2内含子区域,从而制备出一个含有抗性基因的CCR5Δ32打靶载体(图1,step3)。
1 材料与方法
1.1 材料
细菌内同源重组试剂盒购自Gene Bridges公司;BAC克隆(克隆号RPCI 24F11)购自Invitrogen公司;质粒pL452由南京大学模式动物所惠赠;引物购自上海生工公司;氯霉素、四环素、卡那霉素和链霉素购自Amresco公司;L-阿拉伯糖购自Calbiochem公司;Taq酶、pfu高保真酶购自TaKaRa公司;质粒提取试剂盒、凝胶回收试剂盒购自Omiga公司。PCR扩增仪(Epperdorrf公司);凝胶成像仪、电泳仪(Bio-Rad公司);电转化仪(BTX公司)。
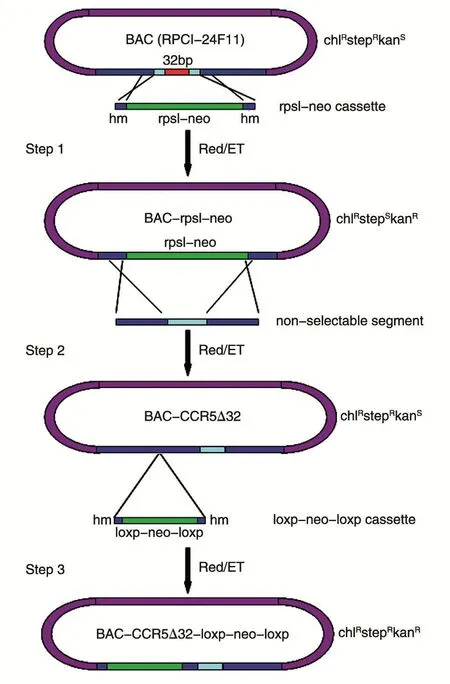
图1 BAC-CCR5Δ32-loxp-neo-loxp载体构建示意图
1.2 含有CCR5同源臂的rpsl-neo片段插入BAC的CCR5基因中
基于同源重组试剂盒提供的rpsl-neo模板,PCR扩增含有CCR5Δ32区域两侧同源臂的rpslneo片段,引物为5'-AATCATCTTTACCAGATCTC AAAAAGAAGGTCTTCATTACACCTGCAGCT/GGCC TGGTGATGATGGCGGGATCG-3'和5'-AGCAGAT GACCATGACAAGCAGCGGCAGGACCAGCCCCAAG ATGACTATC/CTCGAGTCAGAAGAACTCGTCAAGA AGG-3',其中“/”代表5'端同源臂区与 3'端 rpslneo引物的连接处。反应条件:95℃ 5min;95℃1min,58℃ 1min,72℃ 2.5min,30 次循环;72℃ 10min。电泳毕,切割1319bp片段并纯化,然后在用0.3%~0.4%L-阿拉伯糖于37℃诱导45~60min的情况下将此纯化片段电转导入含有BAC和pRedET质粒的大肠杆菌DH10B中,按照后面提及的同源重组步骤进行rpsl-neo插入,以替换含Δ32区域的52bp区,收集细菌涂布于含氯霉素(15μg/mL)、卡那霉素(15μg/mL)、四环素(3μg/mL)的LB琼脂平板上,阳性克隆经鉴定可获得BAC-rpsl-neo。
1.3 180bp非选择性片段插入替换rpsl-neo片段获得CCR5Δ32基因型
首先依据CCR5的Δ32区两侧序列设计并合成2段各100bp的overlap单链DNA片段,该2片段的3'端20bp为碱基互补配对区,它们的碱基序列为5'-ACTTGGGTGGTGGCTGTGTTTGCGTCT CTCCCAGGAATCATCTTTACCAGATCTCAAAAAGA AGGTCTTCATTACACCTGCAGCTCTCATTTTCCATA CA-3'和5'-AGCAGAGTTTTTAGGATTCCCGAGTA GCAGATGACCATGACAAGCAGCGGCAGGACCAGC CCCAAGATGACTATCTTTAATGTATGGAAAATGAG AGCTG-3'。然后直接退火合成180bp的不含Δ 32之32bp的非选择性片段。退火条件:2片段各取 300ng溶于 50μL PCR 反应体系中;94℃4min,以 94℃ 30s、55℃ 45s、72℃ 1min行 35次循环,72℃ 5min。电泳毕,切割180bp片段并纯化,然后同上进行同源重组,将此短片段电转导入含有BAC-rpsl-neo及pRedET质粒的大肠杆菌DH10B,按照后面提及的同源重组步骤进行插入,以替换掉rpsl-neo,收集细菌涂布于含氯霉素(15μg/mL)、卡那霉素(15μg/mL)、四环素(3μg/mL)的LB琼脂平板上(由于BAC上rpsl基因被替换,所以此时菌株在卡那霉素抗性消失的同时具备链霉素抗性),阳性克隆经鉴定可获得BAC-CCR5Δ32。
1.4 扩增loxp-neo-loxp抗性基因片段插入BACCCR5Δ32中
利用在线转录因子预测软件TFSEARCH(http://www.cbrc.jp/papia/howt ouse/howtouse_tfsearch.html)对CCR5所有内含子区域进行转录因子预测,选取第2内含子区的无转录因子段作为插入loxp-neo-loxp片段的靶位点。以质粒pL452为模板,PCR扩增含有CCR5第二内含子同源臂的loxp-neo-loxp抗性基因片段,引物为5'-AAATCT TGAAATCTTATCTTCTGCTAAGGAGAACTAAACCC TCTCCAGTGAGATGCCTTCTGAATATGTGCCCACA AGAA/GAATTCCTGCAGCCCAATTCCGATC-3'和5'-TTAATATTAATTTTGACCATTTTTAGGCTTCCC TCTTGTCTGGAGGAAAAAGAAAAAAGAGAACCAG ACTTAGACACAAC/GCTCTAGAACTAGTGGATCCC CTCG-3',其中“/”代表5'端同源臂区与 3'端 loxpneo-loxp引物的连接处。反应条件:95℃ 5min;98℃ 15s,55℃ 15s,72℃ 2min,5次循环 ;98℃ 15s,65℃ 15s,72℃ 2min,26 次循环;72℃ 5min。电泳毕,切割1909bp片段并纯化,同上电转导入含有BAC-CCR5Δ32及pRedET质粒的大肠杆菌DH10B,按照后面提及的同源重组步骤插入loxp-neo-loxp,收集细菌涂布于含有氯霉素(15μg/mL)、卡那霉素(15μg/mL)的LB琼脂平板上,阳性克隆经鉴定可获得BAC-CCR5Δ32-loxp-neo-loxp。
1.5 电转感受态细菌制备
取50~100μL含有BAC和/或pRedET质粒的DH10B菌液加入4mL携有相应抗生素的LB培养基中,37℃过夜增菌,当D600nm值为0.5~0.8时将菌液置于冰上20~30min,然后分装入2~3支EP管中,4℃、3500r/min离心5min,弃上清,收集细胞沉淀,用冰预冷的10%甘油洗涤3次,按50~80μL/支用10%甘油重悬并置于-80℃保存,临用时解冻即可。
1.6 电击转化与阳性克隆的筛选
在本实验中,BAC克隆需要转化入pRedET和插入片段,其电转化程序完全相同。接种BAC克隆于2mL LB培养基中,并添加15μg/mL氯霉素,37℃、250r/min过夜培养,次日取 50~100μL过夜菌加入含有氯霉素的LB培养基中,37℃、250r/min培养至D600nm值为0.3左右时加入144μL 10%L-阿拉伯糖(终浓度为0.3%~0.4%),继续培养1h左右直到D600nm值为0.5~0.8,然后按照1.5所述方法制备电转感受态细菌用于后续质粒或插入片段的电转(仅电转pRedET质粒而不进行同源重组的话则不需要加入阿拉伯糖诱导这一步,直接进行下面的电转程序即可)。
将制备好的电转感受态细菌混合pRedET质粒和/或rpsl-neo(或180bp非选择性片段,或loxp-neo-loxp),转入电击杯并放入电转仪中,按参数1750 V、5~6 ms进行电转,取出菌液转入一个15mL离心管中,37℃、70min振摇,以诱导同源重组反应的发生。最后将共约100μL菌液涂布于含有特定抗生素的LB琼脂平板上,30℃培养20h,次日挑取菌落进行PCR鉴定,阳性菌落者送测序验证。
2 结果
2.1 BAC-CCR5Δ32的构建
首先PCR扩增rpsl-neo片段后纯化得到1319bp片段(图2A),此片段两端各含有50bp的同源臂,随后将其电转入携有pRedET质粒的BAC菌株(该BAC上含有CCR5基因),在阿拉伯糖的诱导作用下pRedET质粒发挥其同源重组功能,将具有50bp同源臂的rpsl-neo片段准确插入CCR5上的对应区域,琼脂平板阳性菌落可在氯霉素、卡那霉素和四环素都存在的情况下生长,随后阳性菌株的PCR鉴定可见6个克隆均存在1952和685bp片段,表明每个阳性菌株里面的部分BAC发生了正确的同源重组,即rpsl-neo准确插入了相应位置(图2B),此为BAC-CCR5-rpsl-neo。
随后我们将合成的2条寡核苷酸链退火延伸合成了180bp的非选择性片段(图2C),该片段具有rpsl-neo插入位置两侧的相对应的同源臂,并且此片段与该位置的CCR5基因序列完全一致,只是没有Δ32区的32bp碱基序列;然后将此非选择性片段电转入携有pRedET质粒的BACCCR5-rpsl-neo菌株,按照同样的重组程序替换掉rpsl-neo,可获得BAC-CCR5Δ32。构建成功的重组子可在氯霉素、链霉素和四环素都存在的情况下生长,令人惊讶的是随后阳性菌株的PCR鉴定可见3个克隆均为168bp片段,表明每个阳性菌株里面的所有BAC发生了正确的同源重组即不仅rpsl-neo被替换,同时野生型BAC也被非选择性片段置换(图2D)。将BAC-CCR5Δ32菌株提取BAC后送测序,结果表明CCR5成功删除了32bp(图4A)。
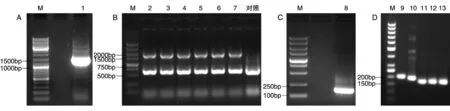
图2 BAC-CCR5Δ32的构建
2.2 BAC-CCR5Δ32-loxp-neo-loxp的构建
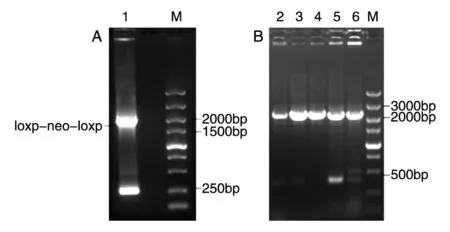
图3 BAC-CCR5Δ32-loxp-neo-loxp的构建
以质粒pL452为模板,PCR扩增含有CCR5第2内含子50bp同源臂的loxp-neo-loxp抗性基因片段,电泳结果表明存在1909bp片段(图3A),切胶纯化后将其电转入BAC-CCR5Δ32菌株,以同样的重组程序获得重组子,该重组子可在氯霉素、卡那霉素和四环素都存在的情况下生长,随后阳性菌株的PCR鉴定可见5个克隆均发生了重组插入,但是只有菌株4显示2条片段(2324和415bp),这表明除4号菌株外其他阳性菌株里的所有BAC发生了正确的同源重组,即loxp-neoloxp准确插入了相应位置(图3B),最后提取BAC送测序,结果表明loxp-neo-loxp成功插入相应位置(图4B),构建了BAC-CCR5Δ32-loxp-neo-loxp载体。
3 讨论
细菌内同源重组技术可在BAC上高效构建各类载体,包括基因打靶载体,尤其适用于受精卵注射打靶载体获得转基因动物。本研究中我们详细阐述了利用基于BAC的细菌内同源重组构建新型的CCR5Δ32打靶载体。首先我们合成了一段2kb左右的rpsl-neo片段,然后重组替换掉一段跨Δ32区域的CCR5上52bp序列,rpsl-neo的特性是赋予了宿主菌卡那霉素抗性和链霉素敏感性(BAC的宿主菌DH10B本身具有链霉素抗性,但rpsl的插入导致其抗性消失);随后我们构建了180bp与CCR5Δ32基因型完全一致的非选择性片段,并利用同样技术重组替换掉rpsl-neo,此时含有BAC-CCR5Δ32的DH10B菌失去了卡那霉素抗性,但恢复了链霉素抗性,通过这种抗性的转换我们筛选到BAC-CCR5Δ32的阳性菌株;最后我们合成并在CCR5内含子区域插入了loxpneo-loxp片段,以赋予该打靶载体卡那霉素和G418抗性,方便将来用于细胞基因打靶的阳性克隆细胞富集。我们选择在内含子区域插入条件性抗性基因片段,目的是将来靶细胞一旦成功地敲除了32bp,获得CCR5Δ32基因型后,可以引入Cre酶的表达去除此外源性的neo抗性基因,残留的一个loxP序列也仅存在于内含子区域,而且事先我们利用软件预测了此内含子区域不存在启动子序列,从而不会影响CCR5Δ32基因后续的表达或表达效率,因此这成为该研究的一个重要的创新之处。
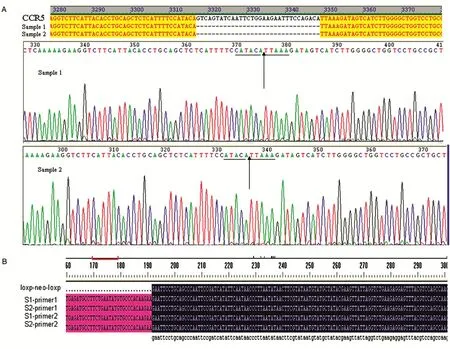
图4 CCR5Δ32和loxp-neo-loxp构建的测序结果
另有一项基于大肠杆菌DY380的同源重组技术,原理与本研究使用的系统基本一样,但是由于它发挥功能必须在DY380细菌中,因此必须将BAC提取后转化入DY380才能进行载体构建,相对而言基于DH10B和pRedET质粒的重组体系就十分便于编辑改造BAC并获得打靶载体[10]。
图2B结果显示所有阳性菌落中rpsl-neo仅把每个阳性菌中的部分而非全部BAC重组置换,而图2D则显示当这些阳性菌在180bp非选择性片段的重组反应中获得一个令人十分惊奇的结果,即不仅类似图2B中的已发生第一次重组反应的BAC-CCR5-rpsl-neo被置换成 BAC-CCR5Δ32,而且同一菌中的未发生第一次重组反应的野生型的BAC也被成功地置换成BAC-CCR5Δ32;同样令人惊奇的,图3B显示5个阳性菌株里有4个的所有BAC都成功地重组插入了loxp-neo-loxp。对于上述两个现象,我们推测可能是由于它们的同源臂区域较rpsl-neo的长所致[18-19],因为非选择性片段的同源臂左右平均各为90bp,loxp-neo-loxp左右同源臂均为80bp,而rpsl-neo却仅为20bp。
总之,我们构建了CCR5Δ32的新型打靶载体,为将来利用此载体在人胚胎干细胞或人血液T细胞中基因打靶获得具有临床意义的CCR5Δ32基因型的hESCs或T细胞奠定了基础。
[1] Deng H,Liu R,Ellmeier W,et al.Identification of a major co-receptor for primary isolates of HIV-1[J].Na⁃ture,1996,381(6584):661-666.
[2] Hutter G,Nowak D,Mossner M,et al.Long-term con⁃trol of HIV by CCR5 Delta32/Delta32 stem-cell trans⁃plantation[J].N Engl J Med,2009,360(7):692-698.
[3] Swan C H,Buhler B,Steinberger P,et al.T-cell pro⁃tection and enrichment through lentiviral CCR5 intra⁃body gene delivery[J].Gene Ther,2006,13(20):1480-1492.
[4] Akkina R,Banerjea A,Bai J,et al.siRNAs,ribo⁃zymes and RNA decoys in modeling stem cell-based gene therapy for HIV/AIDS[J].Anticancer Res,2003,23(3A):1997-2005.
[5] DiGiusto D L,Krishnan A,Li L,et al.RNA-based gene therapy for HIV with lentiviral vector-modified CD34+cells in patients undergoing transplantation for AIDS-related lymphoma[J].Sci Transl Med,2010,2(36):36ra43.
[6] Li L,Krymskaya L,Wang J,et al.Genomic editing of the HIV-1 coreceptor CCR5 in adult hematopoietic stem and progenitor cells using zinc finger nucleases[J].Mol Ther,2013,21(6):1259-1269.
[7] Maier D A,Brennan A L,Jiang S,et al.Efficient clinical scale gene modification via zinc finger nucle⁃ase-targeted disruption of the HIV co-receptor CCR5[J].Hum Gene Ther,2013,24(3):245-258.
[8] Mandal P K,Ferreira L M,Collins R,et al.Efficient ablation of genes in human hematopoietic stem and ef⁃fectorcells using CRISPR/Cas9[J].CellStem Cell,2014,15(5):643-652.
[9] Ye L,Wang J,Beyer A I,et al.Seamless modifica⁃tion of wild-type induced pluripotent stem cells to the natural CCR5Delta32 mutation confers resistance to HIV infection[J].Proc Natl Acad Sci USA,2014,111(26):9591-9596.
[10]Liu P,Jenkins N A,Copeland N G.A highly effi⁃cient recombineering-based method for generating con⁃ditional knockout mutations[J].Genome Res,2003,13(3):476-484.
[11]Sharan S K,Thomason L C,Kuznetsov S G,et al.Re⁃combineering:a homologous recombination-based meth⁃od of genetic engineering[J].Nat Protoc,2009,4(2):206-223.
[12]Bird A W,Erler A,Fu J,et al.High-efficiency coun⁃terselection recombineering for site-directed mutagene⁃sis in bacterial artificial chromosomes[J].Nat Methods,2012,9(1):103-109.
[13]Westenberg M,Soedling H M,Mann D A,et al.Coun⁃ter-selection recombineering ofthe baculovirusge⁃nome:a strategy for seamless modification of repeatcontaining BACs[J].Nucleic Acids Res,2010,38(16):e166.
[14]Wu S,Ying G,Wu Q,et al.A protocol for construct⁃ing gene targeting vectors:generating knockout mice forthe cadherin family and beyond[J].NatProtoc,2008,3(6):1056-1076.
[15]Zhang Y,Buchholz F,Muyrers J P,et al.A new log⁃ic for DNA engineering using recombination in Esche⁃richia coli[J].Nat Genet,1998,20(2):123-128.
[16]Heermann R,Zeppenfeld T,Jung K.Simple genera⁃tion of site-directed point mutations in the Escherich⁃ia coli chromosome using Red(R)/ET(R)recombination[J].Microb Cell Fact,2008,7:14.
[17]Wang S,Zhao Y,Leiby M,et al.A new positive/nega⁃tive selection scheme for precise BAC recombineering[J].Mol Biotechnol,2009,42(1):110-116.
[18]FujimotoR,OsakabeT,SaitoM,etal.Minimum length of homology arms required for effective Red/ET recombination[J].BiosciBiotechnolBiochem,2009,73(12):2783-2786.
[19]Bao Z,Cartinhour S,Swingle B.Substrate and target sequence length influence RecTE(Psy)recombineering efficiency in Pseudomonas syringae[J]. PLoS One,2012,7(11):e50617.
Construction ofCCR5Δ32TargetingVectorwith Red/ET Homologous Recombination
LU Zhi-Yong,YANG Zhuo-Shun,DING Yan,YUAN Ya-Hong,WANG Xiao-Li,YU Li,LI Dong-Sheng*
Life Research Institute,Taihe Hospital,Hubei University of Medicine,Shiyan 442000,China*Corresponding author,E-mail:dsli1698@126.com
Objective:Recently modification of CCR5 locus for anti-HIV-1 infection was performed by the ge⁃nome editing methods like TELENs or CRISPR-Cas with the strategies of disruption of CCR5.To obtain the exact mutation of Δ32 deletion at the locus,we employed a novel strategy for developing CCR5Δ32 targeting vector based on Red/ET homologous recombination.Methods:Firstly,a rpsl-neo cassette flanked by two homology arms to CCR5 was amplified by PCR and then electroporated into E.coli DH10B with the BAC bearing CCR5.Through Red/ET homologous recombination,a 52bp DNA sequences containing Δ32 in CCR5 was replaced by rpsl-neo.Subsequently,a non-selectable fragment was electroporated and replaced the rpsl-neo resulting in Δ32 deletion at CCR5.Furthermore,a loxp-neo-loxp cassette was also introduced into intron 2 within CCR5 through the same machnism,which allows for G418 selection for future targeting human cells and will be cut under the Cre protein only leaving one loxp at the intron region.Results:rpsl-neo cassette was successfully inserted into CCR5 on BAC and replaced by non-selectable fragment and at the same time a loxp-neo-loxp cassette was also introduced into intron 2 of CCR5.Conclusion:CCR5Δ32-loxp-neo-loxp targeting vector was accessed by Red/ET homologous re⁃combination.
CCR5;bacterial artificial chromosome;homologous recombination;recombineering
Q78
A
1009-0002(2017)04-0415-07
2017-02-20
国家科技重大专项(2013ZX10001004-002-005);湖北省教育厅科研项目(B2013111)
卢智勇(1973- ),男,博士,(E-mail)zylever@126.com
李东升,(E-mail)dsli1698@126.com
10.3969/j.issn.1009-0002.2017.04.003
