聚乳酸/PBAT合金的微孔发泡行为研究
2017-11-01余科松周洪福密建国王向东
余科松,周洪福,密建国,王向东*
(1.北京工商大学材料与机械工程学院,北京 100048;2.北京化工大学化学工程学院,北京 100029)
聚乳酸/PBAT合金的微孔发泡行为研究
余科松1,周洪福1,密建国2,王向东1*
(1.北京工商大学材料与机械工程学院,北京 100048;2.北京化工大学化学工程学院,北京 100029)
通过熔融共混法制备了聚乳酸(PLA)/聚己二酸/对苯二甲酸丁二酯(PBAT)合金,同时添加扩链剂和成核剂苯基磷酸锌(PPZn)调整PLA的结晶和发泡性能。动态流变和差示扫描量热分析的研究结果表明,扩链剂可以改善PLA的熔体强度,PPZn可以促进PLA结晶;在实验条件下经过直接降压法发泡后,所制得的PLA/PBAT合金泡沫的泡孔直径为1~4 μm,泡孔密度为109~1011个/cm3,均为微孔泡沫。
聚乳酸;聚己二酸/对苯二甲酸丁二酯;结晶行为;微孔泡沫;发泡行为
0 前言
微孔泡沫最早由Suh等人在19世纪80年代提出[1],根据定义其泡孔尺寸介于1~10 μm,泡孔密度介于109~1012个/cm3[2]。微孔泡沫不仅具有一般泡沫的质轻、比强度高等特点,还具有更优异的力学性能,从而拓展泡沫的应用范围[3]。PLA是一种新型的生物降解材料,但其可发性较差,如何制备PLA的微孔泡沫的制备成为了近年来的研究重点[4]。
本文将PLA和PBAT共混,同时添加成核剂PPZn改善PLA结晶行为,以及扩链剂CE改善其发泡行为,研究PLA/PBAT合金微孔泡沫的形成条件及其发泡行为。
1 实验部分
1.1 主要原料
PLA,2003D,美国Natureworks公司;
PBAT,C1200,德国BASF公司;
PPZn,自制;
扩链剂母粒(含10 %的环氧基低聚物)(CE),Joncryl 4360,德国BASF公司;
CO2,纯度>99 %,市售。
1.2 主要设备及仪器
密炼机,XSS-300,上海科创橡塑机械设备有限公司;
鼓风干燥箱,DHG-9245,上海一恒科技有限公司;
差示扫描量热仪(DSC),Q100,美国TA公司;
旋转流变仪,ARES,美国TA公司;
扫描电子显微镜(SEM),TM-3000,日本Hitachi公司;
真密度计/开闭孔率测定仪,ULTRAPYC,美国Quantachrome公司;
超临界CO2间歇发泡装置,自制。
1.3 样品制备
将实验原料在60 ℃干燥箱中干燥6 h,以排除原料中水分的影响,然后按照表1中所示配比在密炼机中进行共混,密炼温度190 ℃,时间10 min,密炼转速60 r/min;密炼后将样品在190 ℃下压成2 mm厚的薄片储存备用;
发泡实验采用间歇式高压釜发泡,使用超临界CO2作为发泡剂;将样品密封在釜内,在10 MPa下选取不同温度(110、120、130 ℃),恒温恒压5 h等CO2充分溶解于样品后,采用直接降压法快速释放釜内压力至常压发泡成型,从而制得泡沫样品;泡沫放置数天熟化,从中随机抽取试样用于进一步测试。

表1 PLA/PBAT共混物实验配方Tab.1 Experimental formula of PLA/PBAT
1.4 性能测试与结构表征
动态流变分析:采用平行板模具,样品直径20 mm,间距1.0 mm,角速度范围0.1~100 rad/s,温度190 ℃,应变保持在10 %以确保在线性黏弹区范围内测试样品的流变性能;
DSC分析:采用氮气气氛,快速升温到190 ℃,保持5 min消除热历史,之后以10 ℃/min的冷却速率降温至40 ℃得到降温结晶曲线,再以10 ℃/min的加热速率升温至190 ℃,得到升温熔融曲线,PLA的结晶度可以通过式(1)计算:
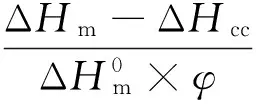
(1)
式中Xc——PLA的绝对结晶度, %
ΔHm——样品的熔融焓,J/g
ΔHcc——样品的冷结晶焓,J/g
φ——共混体系中PLA的质量分数
泡孔形态表征测试:将样品在液氮中冷冻脆断并在断面喷金后,在SEM下观察泡孔结构并计算泡孔密度,泡孔密度通过计算机软件Image-Pro Plus分析,并通过式(2)和式(3)计算[5]:
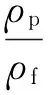
(2)
(3)
式中φ——发泡倍率
N——泡孔密度,个/cm3
n——统计面积中泡孔数量,个
M——SEM照片放大倍数
A——SEM照片中所选择的统计面积,cm2
ρp——发泡前样品密度,g/cm3
ρf——发泡后的样品密度,g/cm3
2 结果与讨论
2.1PLA/PBAT/成核剂/扩链剂的动态流变性能

■—PLA ●—PLA/PBAT ▲—PLA/PBAT/CE ▼—PLA/PBAT/PPZn ◆—PLA/PBAT/CE/PPZn图1 样品的G′与角速度的关系Fig.1 G′ of the samples against ω
对发泡过程来说,熔体强度是需要首要考虑的因素之一。在气泡增长阶段如果熔体强度过低会导致气泡的破裂及气体的逸散,这往往会使得泡孔塌陷而无法形成均匀的泡沫制品。通过动态流变性能可以获得聚合物熔体的黏弹性,从而表征其熔体强度。储能模量(G′)可以用于描述聚合物的“可发性”,G′越大则熔体强度越高[6]。在低频区,PLA的储能模量较低,在PLA中加入PBAT后,G′有了显著的提高,说明PBAT可以提高PLA的熔体强度,改善其可发性。PLA/PBAT的共混体系中加入PPZn后G′变化不大,而加入CE后G′增加。这是因为PPZn作为PLA的结晶成核剂,添加量不高且在动态流变的测量条件下(190 ℃)体系并没有晶体存在所以对共混体系不能产生明显的影响;而CE作为PLA的扩链剂,可以使PLA产生支化结构,这些支链可以形成物理缠结从而提高聚合物基体的熔体强度。
2.2 PLA及PLA/PBAT合金的结晶性能分析

(a)PLA (b)PLA/PBAT (c)PLA/PBAT/CE (d)PLA/PBAT/PPZn (e)PLA/PBAT/CE/PPZn图3 未发泡PLA、PBAT以及在PLA/PBAT共混物中添加CE或PPZn的样品的断面SEM照片Fig.3 SEM of fracture surfaces of unfoamed PLA, PBAT and their blends with the addition of CE and PPZn
发泡过程中聚合物所含晶体的数量会对泡孔直径及发泡倍率产生影响。这是因为结晶的存在可以提供相界面,相界面有利于CO2的富集,作为气泡成核点。大量的气泡成核点是得到微孔泡沫结构的关键因素之一。从图2中可以看到,PLA在118 ℃存在冷结晶峰,其晶体的形成基本都集中在升温的冷结晶过程中而降温过程并没有明显的结晶峰(图未列出),在147 ℃存在熔融峰,其结晶度为19.6 %,而纯PBAT的熔融曲线表现为121 ℃附近一个宽泛的熔融峰。PLA中加入PBAT之后,冷结晶峰变得不再明显,且PLA结晶度下降到不足5 %,说明PBAT对PLA的结晶是有阻碍作用的,其原因可能是不相容的PBAT分散于PLA中破坏了PLA的有序性使之更难形成晶体。在PLA/PBAT的共混物中加入扩链剂CE后,PLA的结晶度进一步下降,这是由于CE促使PLA产生支化结构,而支化结构使得PLA链段更加无序,且经过支化的PLA可以起到物理缠结的作用阻碍分子运动。在共混物中加入PPZn则会使得PLA的结晶度显著提高,回到17.7 %左右,且使得冷结晶温度下降5 ℃左右。这是因为PPZn作为PLA的成核剂可以促进PLA的结晶,使得冷结晶可以在更低温度下形成。

1—PLA 2—PBAT 3—PLA/PBAT 4—PLA/PBAT/CE 5—PLA/PPZn 6—PLA/PBAT/CE/PPZn图2 PLA、PBAT以及在PLA/PBAT共混物中添加 CE或PPZn的DSC曲线Fig.2 DSC curves of PLA, PBAT and their blends with the addition of CE and PPZn
2.3 发泡前样品断面形貌表征
从图3(a)中可以看到,PLA样品的断面存在大量的裂纹,这是由于PLA的脆性而在制样过程中形成的。加入PBAT后,裂纹明显减少,可以看到PBAT在PLA中以“海岛结构”均匀分散开,PLA为连续相(即“海”),PBAT呈直径1.4 μm左右的小球为分散相(即“岛”)。这是因为PBAT和PLA的相容性并不好[7],共混后形成非均相体系。这样使得PLA和PBAT之间存在大量的相界面,这对于发泡过程的气泡成核以及增加CO2的溶解是有利的。从图3(d)、(e)中可以看到,成核剂PPZn(黑色箭头所指的白色不规则颗粒)分散在PLA相中。在加入PPZn之后,PBAT形成的小球变得更少,而且在图3(e)中,小球有合并成大球的趋势。
2.4 发泡样品
在微孔泡沫的发泡成型过程中,瞬间产生大量的气泡成核点,以及及时控制气泡增长是整个工艺的核心。后者可以通过控制熔体强度和结晶等来控制,这在前面已经提到;另一方面要使得体系产生大量气泡成核点则需要大量的相界面以及足够的CO2溶解度[8]。为使得CO2能够充分溶解在聚合物基体中,本文的发泡过程采用10 MPa下用超临界CO2浸泡5 h,发泡温度选取110、120、130 ℃。

样品:(a)、(e)、(i)PLA/PBAT (b)、(f)、(j)PLA/PBAT/CE (c)、(g)、(k)PLA/PBAT/PPZn (d)、(h)、(l)PLA/PBAT/CE/PPZn 温度/℃:(a)、(b)、(c)、(d)130 (e)、(f)、(g)、(h)120 (i)、(j)、(k)、(l)110图4 不同浸泡温度下发泡样品的SEM照片Fig.4 SEM of different samples foamed at various temperature
图4是在不同温度下发泡后样品的泡孔形貌图,横向为同一发泡温度,纵向为同一样品。表2是根据密度测量及式(2)、式(3)计算得出的泡孔直径、泡孔密度以及发泡倍率。结合图4和表2的内容可以看到,在实验条件下所制得的泡沫,其泡孔直径为1~4 μm,泡孔密度109~1011个/cm3,均达到了微孔泡沫的级别。当温度较高时,发泡倍率更大,泡孔密度也更高。可以看到当发泡温度减少到110 ℃时,单纯的PLA/PBAT共混样品的泡沫,其泡孔结构已经无法形成连续的分布,而是呈片的分布在一些分散的区域中,泡孔密度也较小这是由于该温度下PBAT只有少部分熔融,而PLA中也有较多部分已经完成冷结晶,晶区的存在阻碍了CO2的溶解和扩散,在这些区域就很难形成气泡。另一方面可以发现在部分泡孔中还存在着小颗粒,结合前面的研究可以知道这些小颗粒就是PBAT的颗粒。由此可以证明发泡过程中有一部分气体在PBAT颗粒周围聚集从而形成泡孔。当向PLA/PBAT的共混体系中加入CE扩链剂或者成核剂PPZn后,泡孔密度得到增加。这是因为当加入扩链剂后聚合物的自由体积增加,有利于CO2的溶解扩散;而成核剂的加入有利于PLA形成小尺寸的结晶,从而减少成片的晶区,这对CO2的溶解扩散也是有利的。并且可以看到,加入CE对泡孔密度的增加作用要好于加PPZn。
结合DSC曲线可知,在120~130 ℃之间,PLA由于冷结晶的作用而结晶,这些晶体不仅可以作为气泡成核点促进气体的聚集使泡沫具有较大的泡孔密度,同时也可以限制气体的逸散以及泡孔的增长,使泡孔直径限制在较小的范围。另一方面,在120~130 ℃之间PBAT大部分处于黏流态,PLA和PBAT的不相容性使得气体更容易在两者的相界面之间聚集。当降压过程发生时,气体先是在界面处快速汇集,由于PLA此时为橡胶态而PBAT为黏流态,导致气体容易向微米颗粒PBAT中以及PLA的晶体表面聚集扩散形成微米级的小气泡,而PLA晶体的存在又阻碍了气泡的进一步发展,当温度降低后泡孔壁被固定,于是大量的微孔结构被固定下来。
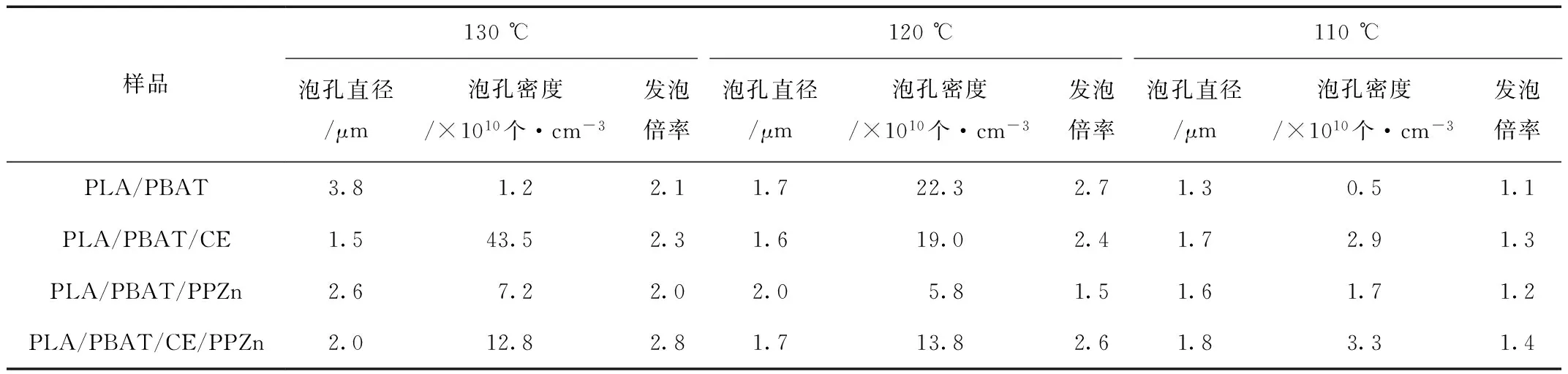
表2 不同发泡温度下发泡样品的泡孔参数Tab.2 Cell parameter of various foamed samples at different temperature
作为对比,图5所示是130 ℃,10 MPa,浸泡5 h的纯PLA发泡样品的SEM照片,可以看到在该温度下PLA难以形成均一的泡孔结构,泡孔结构集中在一定的区域内分布。这是由于在130 ℃时PLA中存在许多的晶体,这些晶体更多的是成片的晶区分布,导致在该实验条件下PLA内溶解的CO2十分有限,所以在发泡过程中只有一些溶解度较高的无定型区域有泡孔产生。PBAT的加入为PLA带来大量的相界面,这些相界面的存在对微孔泡沫的形成起到了重要的作用。

图5 发泡温度为130 ℃时PLA泡沫的SEM照片Fig.5 SEM of PLA foamed at 130 ℃
3 结论
(1)在PLA中引入与之相容性不好的PBAT可以形成“海岛结构”增加两相界面,有利于气泡成核;
(2)PPZn可以促进PLA结晶,形成的晶体一方面可以作为气泡成核点,另一方面抑制发泡过程中的气泡增长过程,而扩链剂CE的加入可以增加聚合物的熔体强度起到约束发泡过程中气体逸散的作用;
(3)PLA/PBAT合金采用直接降压法在120、130 ℃,10 MPa下浸泡5 h都能形成泡孔形态较好的微孔泡沫,而当温度处于110 ℃附近,泡孔将会呈现区域性分布,难以形成均一泡孔。
[1] Martini-Vvedebsky J K, Suh N P, Waldman F A. Microcellular Closed Cell Foams and Their Method of Manufacture: U S,4473665[P]. 1984-09-25.
[2] Waldman F A. The Processing of Microcellular Foam[J]. Massachusetts Institute of Technology, 1982.
[3] 牟文杰, 吴舜英. 微孔泡沫塑料成型技术[J]. 塑料, 2001,(3):33-36.
Mu Wenjie, Wu Shunying.Formation Theory of Microcellular Foam Plastics[J]. Plastics, 2001,(3):33-36.
[4] 杨志云, 蔡业彬, 张铱鈖. PLA泡沫塑料的研究进展[J]. 广东石油化工学院学报, 2014,(3):10-13.
Yang Zhiyun, Cai Yebin, Zhang Yifen. Study on the Photochemical Reaction Character of the Reinecke Salt[J]. Journal of Guangdong University of Petrochemical Technology, 2014,(3):10-13.
[5] Wang X, Liu W, Zhou H, et al. Study on the Effect of Dispersion Phase Morphology on Porous Structure of Poly (Lactic Acid)/Poly (Ethylene Terephthalate Glycol-Modified) Blending Foams[J]. Polymer, 2013, 54(21):5839-5851.
[6] 王向东, 刘本刚, 陈士宏,等. 不同聚丙烯发泡体系的挤出发泡行为研究[J]. 中国塑料, 2006,(11):76-81.
Wang Xiangdong, Liu Bengang, Chen Shihong, et al. Extrusion Foaming Behavior of Different Polypropylene Foaming Systems[J]. China Plastics, 2006,(11):76-81.
[7] 邬昊杰, 赵佳旭, 吴智华. PBAT及增容剂含量对PLA/PBAT共混物结构和性能的影响[J]. 合成树脂及塑料, 2015,(2):5-9.
Wu Haojie, Zhao Jiaxu, Wu Zhihua. Effects of Contents of PBAT and Compatibilizer on Microstructure and Pro-perties of PLA/PBAT Blends[J]. China Synthetic Resin and Plastics, 2015,(2):5-9.
[8] 刘 阳, 史学涛, 张广成,等. PLA及其增韧共混体系超临界CO2发泡行为[J]. 高分子材料科学与工程, 2015, 31(4):61-67.
Liu Yang, Shi Xuetao, Zhang Guangcheng, et al.Foaming Process of Polylactide and Toughened Polylactide Blends Using Supercritical CO2[J]. Polymer Materials Science and Engineering, 2015, 31(4):61-67.
StudyonMicrocellularFoamPropertiesofPLA/PBATBlends
YU Kesong1, ZHOU Hongfu1, MI Jianguo2, WANG Xiangdong1*
(1.School of Materials Science and Engineering, Beijing Technology and Business University, Beijing 100048, China; 2.College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, China)
Poly(lactic acid) (PLA)/poly(butyleneadipate-co-terephthalate) (PBAT) blends were prepared by melt blending with the aid of chain extender (CE) and nucleating agent (PPZn). Dynamic rheological characterization and differential scanning calorimetry analysis indicated that the melt strength and crystallization of PLA were improved by addition of CE and PPZn, respectively. Microcellular foams based on PLA/PBAT blends were prepared under the optimal condition developed by this work, and the foams obtained a cell size of 1~4 μm and a cell density of 109~1011cells/cm3.
poly(lactic acid); poly (butyleneadipate-co-terephthalate); crystallization behavior; microcellular foam; foaming behavior
TQ321
B
1001-9278(2017)10-0055-06
10.19491/j.issn.1001-9278.2017.10.010
2017-07-25
北京市自然科学基金资助项目(2162012)、国家自然科学基金资助项目(51673004)
*联系人,wangxid@th.btbu.edu.cn
