FSD-C10调节阿尔茨海默病双转基因小鼠炎性微环境*
2017-10-20谷青芳尉杰忠李艳花樊慧杰肖保国马存根
谷青芳, 尉杰忠▲ , 吴 昊, 李艳花, 樊慧杰, 柴 智,王 青, 肖保国,3, 马存根,△
(1大同大学脑科学研究所, 山西 大同 037009; 2山西中医学院“2011”协同创新中心/神经生物学研究中心,山西 太原 030024; 3复旦大学华山医院神经病学研究所, 上海 200025)
·论著·
FSD-C10调节阿尔茨海默病双转基因小鼠炎性微环境*
谷青芳1, 尉杰忠1▲, 吴 昊1, 李艳花1, 樊慧杰2, 柴 智2,王 青2, 肖保国1,3, 马存根1,2△
(1大同大学脑科学研究所, 山西 大同 037009;2山西中医学院“2011”协同创新中心/神经生物学研究中心,山西 太原 030024;3复旦大学华山医院神经病学研究所, 上海 200025)
目的探讨新型Rho激酶抑制剂FSD-C10对阿尔茨海默病(Alzheimer disease,AD)模型小鼠脑内炎性微环境的调节作用。方法采用双转染人β-淀粉样蛋白前体(β-amyloid protein precursor,APP)695swe基因和人早老素1(presenilin-1,PS1)ΔE9突变基因的8月龄小鼠作为AD动物模型,随机分为模型组和FSD-C10治疗组,分别经腹腔注射生理盐水和FSD-C10 (25 mg·kg-1·d-1)持续治疗2个月,同月龄野生型小鼠作为正常对照组。应用Morris水迷宫(Morris water maze,MWM)实验检测小鼠学习和记忆能力。采用免疫组化和Western blot技术检测小鼠脑组织β-淀粉样蛋白(Aβ)、磷酸化Tau蛋白(p-Tau)、β位点APP剪切酶(BACE)、Toll样受体4(TLR-4)、磷酸化核因子κB(p-NF-κB)、诱导型一氧化氮合酶(iNOS)和精氨酸酶1(Arg-1)的表达。结果与模型组相比,FSD-C10干预能显著改善APP/PS1双转基因小鼠学习和记忆能力,减少海马区Aβ1-42、p-Tau和BACE的表达,抑制脑内炎症信号通路TLRs/NF-κB轴TLR-4的表达和p-NF-κB的激活,减少iNOS的表达,增加Arg-1的表达。结论FSD-C10干预能明显改善APP/PS1双转基因小鼠的学习和记忆能力,其机制可能是通过抑制TLRs/NF-κB信号通路激活,减少炎症因子的分泌及促进M1型炎性小胶质细胞向M2型抗炎小胶质细胞转化,从而改善APP/PS1双转基因小鼠脑组织炎症微环境。
APP/PS1双转基因小鼠; FSD-C10; 炎症微环境; TLRs/NF-κB通路
根据世界卫生组织最新统计,预测到老龄化程度更为严峻的2050年,全球阿尔茨海默病(Alzheimer disease,AD)患病人数将超过1亿[1]。迄今为止AD尚无特效药物治疗,2014年初辉瑞、强生、礼来公司联合研发的抗Aβ单克隆抗体药物治疗AD的研究在III期临床试验也遭到失败。这些探索也让人们开始重新审视AD的新药研发和治疗策略。
研究发现,Rho相关激酶(Rho-associated kinase,ROCK)的异常激活在AD的病理过程中扮演着重要的角色[2-3]。抑制ROCK的活性有利于防止炎症反应和神经损伤。已有研究证实非甾体类抗炎药通过抑制ROCK的活性降低AD的发病率[4]。因此,抑制ROCK可能是治疗AD的有效策略。我们前期实验已证实ROCK抑制剂fasudil对AD具有良好的治疗效果[5],但其生物利用度差、长期使用的安全窗较小的缺点限制了它的临床应用。新型ROCK抑制剂FSD-C10的效价与fasudil等同,且对神经元毒性小,对血管影响小。基于此,本研究进一步探讨了FSD-C10 对双转染人β-淀粉样蛋白前体(β-amyloid protein precursor,APP)/人早老素1(presenilin-1,PS1)基因的小鼠认知功能及脑组织炎性微环境的作用,为开发新型ROCK抑制剂和临床应用ROCK抑制剂治疗AD提供实验依据。
材 料 和 方 法
1材料
1.1实验动物 实验共分3组,雄性8月龄APP695swe/PS1-ΔE9转基因小鼠16只随机分为模型组与FSD-C10干预组,同窝雄性8 月龄C57BL/6小鼠8只作为正常对照组。上述小鼠体重18~22 g,购自上海南方模式生物科技发展有限公司(动物实验遵循山西大同大学生物医学研究伦理审查委员会制定的实验动物伦理原则执行科学实验)。实验动物置于室温和光照可控的无菌动物房清洁饲养[(25±2) ℃],可自由进食。
1.2主要试剂 BCA蛋白定量试剂盒购自中国碧云天生物技术有限公司;FSD-C10化合物由天津红日药业股份有限公司友好提供。
1.3主要仪器 Morris水迷宫(Morris water maze,MWM)软件SMART V3.0及硬件购自深圳瑞沃德生命科技有限公司;冰冻切片机购自Leica;酶标仪购于THERMO;凝胶成像分析仪购自Bio-Rad;荧光显微镜购自Olympus。
2方法
2.1AD模型鼠的建立APP/PS1双转基因小鼠可表达人APP695swe基因和PS1-ΔE9基因,能产生高水平的Aβ1-42纤维沉积,在8月龄时可出现Aβ老年斑,是国际通用的AD模型鼠之一。
2.2动物分组及给药APP/PS1实验小鼠随机分为模型组与干预组,分别腹腔注射生理盐水(0.9% NaCl,n=8)和FSD-C10(25 mg·kg-1·d-1,n=8),持续给予2 个月,野生组取8只正常老龄小鼠作为对照同法给予等量生理盐水。
2.3水迷宫检测 水迷宫为直径90 cm,高50 cm的圆形水池,装满用食用白色素调和形成的不透明的水,水迷宫上方安置带有显示系统的摄像机,置于一个安静、持续光照的地方,水温保持在23~25 ℃。水迷宫硬件分为4个象限(NW、NE、SW和SE),在SW象限放一个直径5.0 cm的圆形平台,平台置于液面下2 cm。小鼠治疗2个月后进行5 d水迷宫训练,每天的上、下午各1次,训练小鼠从起点到达平台的时间和距离,每次时长60 s,如果在60 s期间小鼠没有到达平台,则进行引导帮助小鼠到达平台,在小鼠到达平台后停留10 s。在训练结束后,进行为期5 d的认知功能测定,然后撤除平台进行空间探索实验判断小鼠对平台空间位置的记忆能力。实验过程中,小鼠游动的踪迹被SMART 3.0系统摄像头跟踪并记录各项指标,包括动物从入水到平台的时间、动物达到平台所在位置的平均距离、动物第1次入水到达平台所在象限(SW区)时间、动物在平台象限滞留时间百分比、动物在平台象限滞留路径百分比和动物在平台象限活动度百分比等。
2.4免疫组化染色观察 行为学测试结束后处死动物,各组随机选取4只小鼠,用4%多聚甲醛灌流进行体内组织固定,然后快速分离脑组织,包埋后行10 μm冠状冰冻切片,切片经PBS洗3次,每次5 min;分别加抗Aβ1-42(1∶500,Millipore)、抗p-Tau(1∶500,Cell Signaling Technology)、抗β位点APP剪切酶(β-site APP-cleaving enzyme,BACE; 1∶500,Cell Signaling Technology)、抗Toll样受体4(Toll-like receptor 4,TLR-4; 1∶500,Cell Signaling Technology)、抗核因子κB (nuclear factor-κB, NF-κB; 1∶500,Cell Signaling Technology);抗诱导型一氧化氮合酶(inducible nitric oxide synthase,iNOS; 1∶500,Cell Signaling Technology)、抗精氨酸酶1(arginase-1,Arg-1; 1∶500,Cell Signaling Technology)及抗CD11b(1∶400,Cell Signaling Technology)4 ℃孵育过夜。次日PBS洗3次;加Alexa Fluor 555或Alexa Fluor 488荧光标记的II 抗(1∶500,Thermo Scientific)室温孵育2 h,50%甘油封片,光镜下观察脑组织海马及皮层区域AD病理标志物,免疫阳性细胞荧光强度,用Image-Pro Plus软件计数并求其均数。
2.5Western blot实验 各组另外4只小鼠冰上快速取脑组织,用组织裂解液在4 ℃条件下裂解组织蛋白,并用BCA法测定蛋白含量,制备蛋白上样缓冲液样品,进行SDS-PAGE蛋白分离。电泳完毕后,用湿式转移法转移到PVDF膜。膜置于5%脱脂牛奶封闭,加入抗Aβ1-42(1∶500)、抗p-Tau(1∶500)、抗BACE(1∶500)、 抗ROCK-II(1∶500)、抗TLR-4(1∶500)、抗p-NF-κB(1∶500)、抗iNOS(1∶500)、抗Arg-1(1∶500)、抗β-actin(1∶500,Epitomics)、抗GAPDH(1∶500,Epitomics)及相应的HRP偶联 II 抗(1∶1 000,Cell Signaling Technology)。使用Bio-Rad凝胶成像分析仪检测染色条带强度,以检测蛋白条带与内参照β-actin或GAPDH的吸光度(A)比值表示。
3统计学处理
采用GraphPad Prism 5.0统计软件进行处理。计量资料用均数±标准差(mean±SD)表示,3组组间比较采用ANOVA及Dunnett’s Post-Hoc检验,2组比较采用t检验,以P<0.05为差异有统计学意义。
结 果
1FSD-C10有效改善APP/PS1双转基因小鼠的学习记忆能力
APP/PS1双转基因小鼠具有明显的记忆能力障碍,为了检测FSD-C10能否改善这种障碍,我们首先通过水迷宫实验对小鼠认知功能进行检测。
定位航行实验统计结果显示,野生组小鼠搜索平台的潜伏期时间明显低于模型组(P<0.05), 小鼠找到平台所经过的平均距离明显低于模型组(P<0.01),小鼠第1次入水到达平台所在象限用的时间明显少于模型组(P<0.01),说明APP/PS1双转基因小鼠具有明显的学习记忆能力的障碍。经过持续 2 个月使用FSD-C10治疗,与模型组比较,FSD-C10干预组的搜索潜伏期明显缩短(P<0.05),找到平台的平均距离(P<0.05)及第1次入水到平台达象限的时间也明显缩短(P<0.05)。
在空间探索实验中,小鼠在目标象限滞留时间百分比和在目标象限游滞留路程百分比干预组均高于模型组小鼠(P<0.05),而3组相比较动物肢体运动功能变化的差异无统计学显著性,可排除肢体功能对运动的影响。结果表明经FSD-C10干预有效改善了APP/PS1双转基因小鼠的学习和记忆能力,见图1。
2FSD-C10减少APP/PS1双转基因小鼠脑内Aβ1-42沉积、p-Tau生成和BACE蛋白表达
Aβ1-42免疫组化染色结果显示,APP/PS1模型组和FSD-C10干预组的海马齿状回(dentate gyrus,DG)区均有Aβ1-42的阳性表达,模型组小鼠脑内海马DG区Aβ1-42的阳性表达数量多、体积大,FSD-C10干预组小鼠脑内海马DG区Aβ1-42的阳性表达数量少、体积小,2组比较差异有统计学显著性(P<0.01)。而野生组小鼠海马DG区未见明显Aβ阳性反应。Western blot实验结果显示,FSD-C10干预组和野生组小鼠脑内的Aβ沉积水平均低于模型组(P<0.01)。
观察p-Tau蛋白免疫组化染色结果显示,模型组和FSD-C10干预组的海马区均有p-Tau 的阳性表达,与模型组比较,FSD-C10干预组小鼠海马区p-Tau 蛋白的阳性数量明显减少(P<0.01)。 而野生组小鼠海马区未见明显p-Tau 蛋白阳性表达。Western blot分析结果显示,FSD-C10干预组和野生组小鼠脑内的p-Tau 蛋白水平均低于模型组。
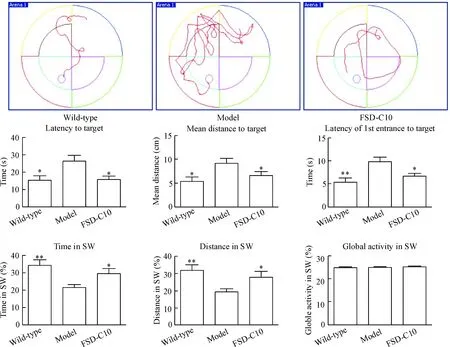
Figure 1. FSD-C10 improved spatial learning ofAPP/PS1 double transgenic mice analyzed by Morris water maze test. Mean±SD.n=8.*P<0.05,**P<0.01vsmodel group.
图13组小鼠水迷宫测试认知功能6项指标的比较
BACE 蛋白免疫组化染色结果显示,与野生组比较,模型组小鼠脑内海马区域BACE蛋白的阳性数明显增加,与模型组比较,FSD-C10干预明显减少BACE蛋白表达(P<0.01)。 Western blot计量结果显示,与模型组相比,FSD-C10干预组小鼠脑内的BACE蛋白水平明显降低(P<0.01),但尚未达到野生组的水平,见图2。
3FSD-C10抑制APP/PS1双转基因小鼠脑组织炎症微环境
炎症反应在AD病理过程中发挥着重要的作用,我们通过免疫组化和Western blot技术检测小鼠脑内海马和皮层区TLRs/NF-κB炎症信号通路相关因子的表达。
免疫组化结果显示,模型组小鼠炎症因子TLR-4和p-NF-κB的阳性细胞数增多,在小鼠脑内海马区和皮质部位均有明显的表达,而FSD-C10干预组TLR-4和p-NF-κB的蛋白水平明显减少,两组差异有统计学意义(P<0.05)。Western blot检测结果发现,TLR-4和p-NF-κB在各组的表达趋势与免疫组化结果一致,与模型组相比,FSD-C10干预组小鼠脑内TLR-4和p-NF-κB的蛋白水平明显低于模型组(P<0.05 和P<0.01);FSD-C10干预组小鼠ROCK-II的表达较模型组降低,差异有统计学意义(P<0.05)。免疫组化和Western blot两种实验方法从定位和定量两方面证明,FSD-C10干预可抑制APP/PS1双转基因小鼠TLRs/NF-κB炎症信号通路的激活,改善炎症微环境,减轻脑内炎症反应,见图3。
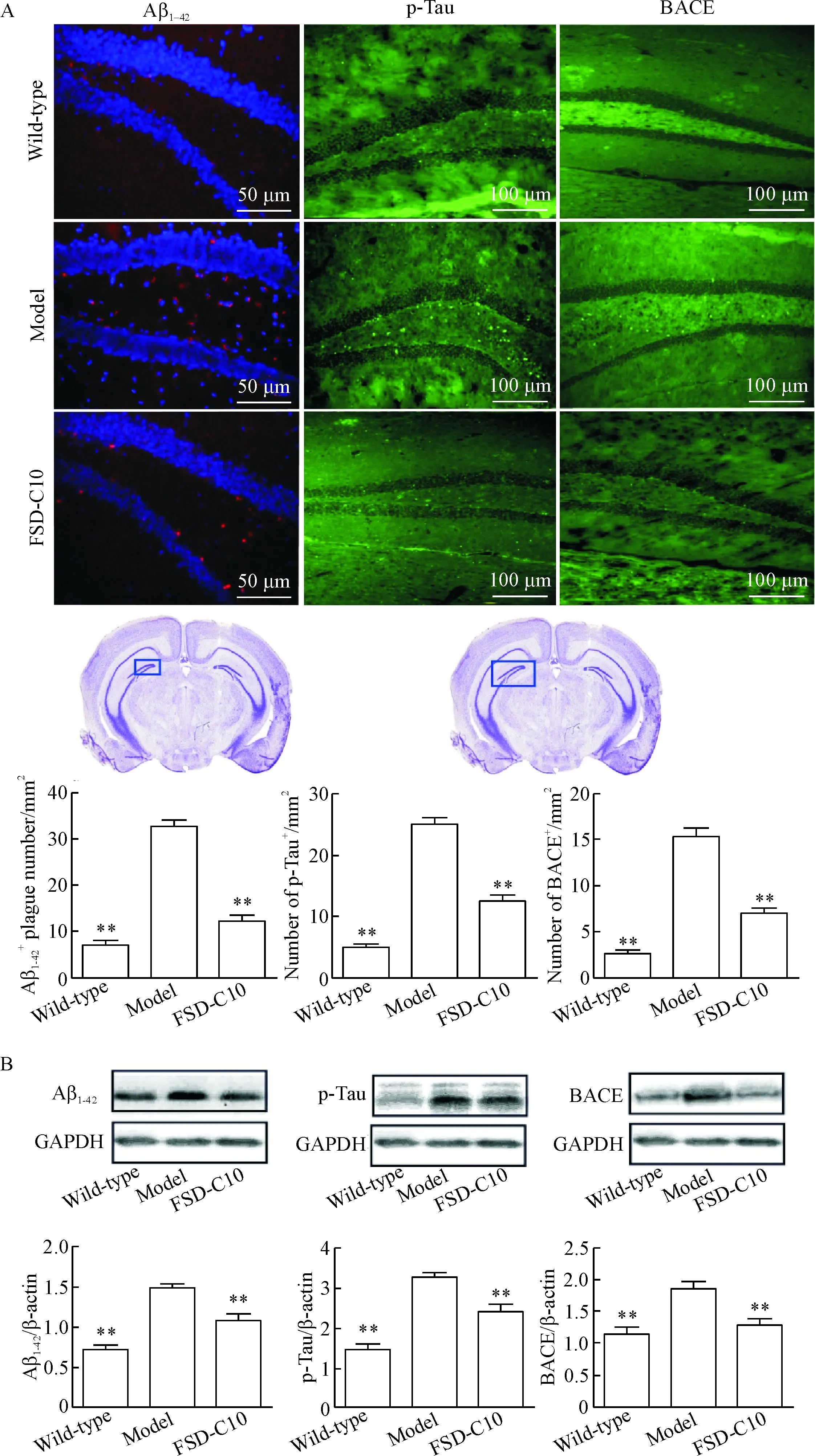
Figure 2. FSD-C10 attenuated Aβ1-42deposition, Tau phosphorylation and BACE expression in theAPP/PS1 double transgenic mice. A: representative photomicrographs and quantitative analysis of Aβ1-42, p-Tau and BACE protein expression in dentate gyrus area of mouse hippocampus observed by immunohistochemical staining; B: the protein levels of Aβ1-42, p-Tau and BACE determined by Western blot. Mean±SD.n=4.**P<0.01vsmodel group.
图23组小鼠病理标志物Aβ1-42、p-Tau和BACE免疫组化染色和Westernblot检测结果的比较
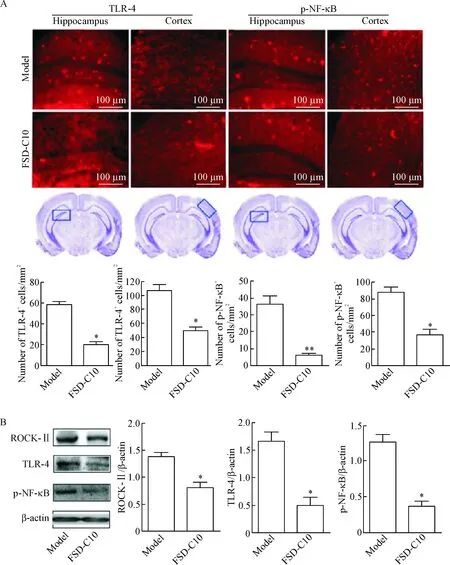
Figure 3. FSD-C10 inhibited inflammatory microenvironment in theAPP/PS1 double transgenic mice. A: representative photomicrographs and quantitative analysis of TLR-4 and p-NF-κB in the hippocampus and cortex of the mice observed by immunohistochemical staining; B: the protein levels of ROCK-II, TLR-4 and p-NF-κB in the brain determined by Western blot. Mean±SD.n=4.*P<0.05,**P<0.01vsmodel group.
图3AD模型组与FSD-C10治疗组小鼠TLRs/NF-κB炎症信号通路免疫组化染色和Westernblot检测结果的比较
4FSD-C10抑制APP/PS1双转基因小鼠脑组织iNOS的表达和增加Arg-1的表达
小胶质细胞(microglia,MG)是 AD 炎症反应中最主要的炎症细胞,小胶质细胞根据功能状态不同分为M1型和M2型。我们进一步通过免疫组化和Western blot技术检测小胶质细胞极化的变化。
iNOS为M1型小胶质细胞标志物,Arg-1为M2型小胶质细胞标志物。免疫组化结果显示,与模型组比较,FSD-C10干预组小鼠海马区iNOS阳性细胞数减少(P<0.05),而Arg-1蛋白阳性的细胞数则增多,差异有统计学意义(P<0.01)。Western blot检测结果发现,iNOS和Arg-1在各组的表达趋势与免疫组化结果一致,FSD-C10 干预组iNOS的表达明显低于模型组(P<0.01),而Arg-1表达则显著高于模型组(P<0.05)。结果提示FSD-C10可抑制iNOS表达,增加Arg-1表达,具有使M1型炎性小胶质细胞向M2型抗炎性细胞转化的效能,起到抗炎和神经保护作用,见图4。
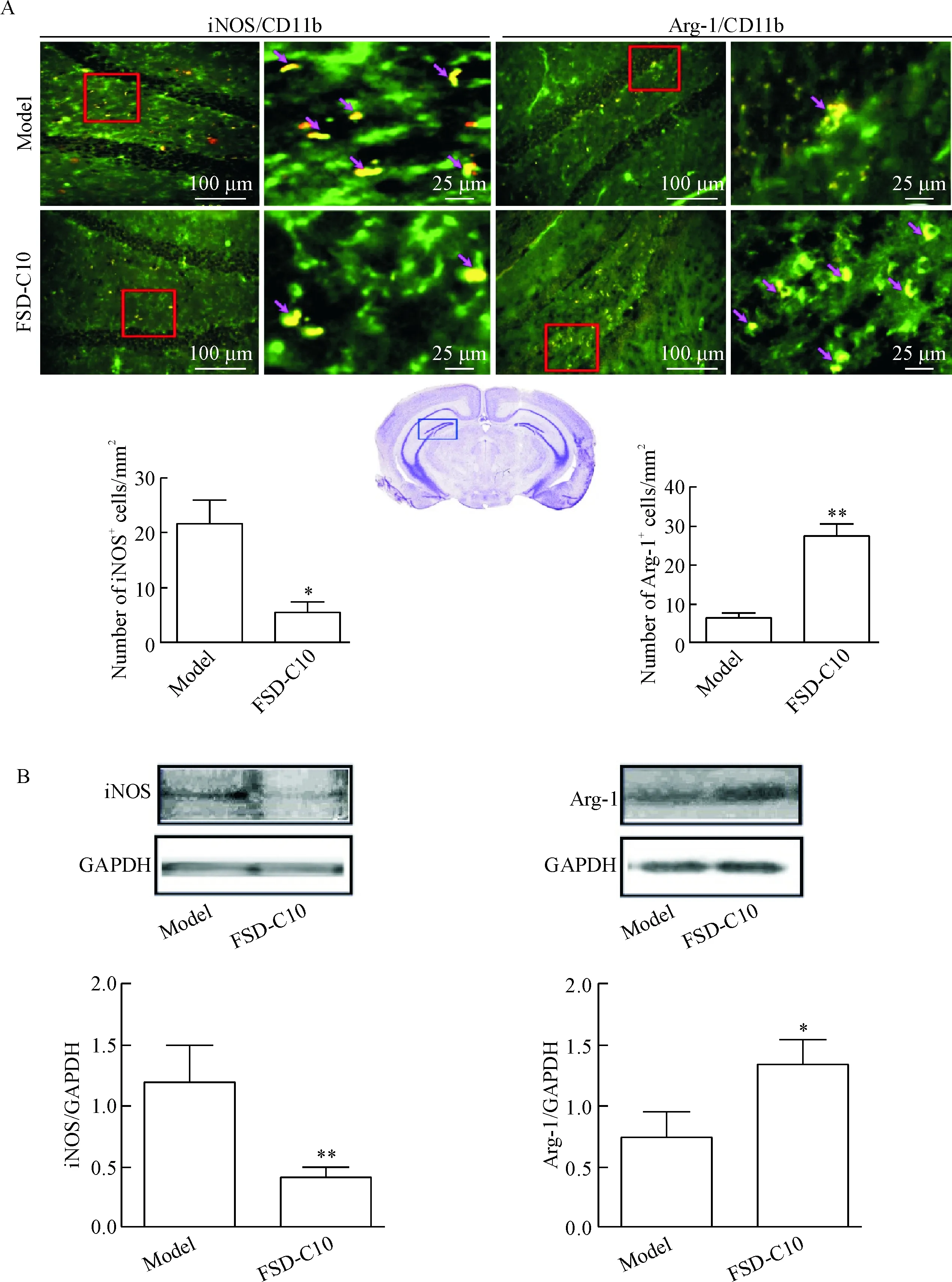
Figure 4. FSD-C10 inhibited the expression of iNOS and increased the expression of Arg-1. A: the double staining of CD11b (green) and iNOS/Arg-1 (red) by immunohistochemical observation; B: representative bands and quantitative analysis of Western blot for determining iNOS and Arg-1. Mean±SD.n=4.*P<0.05,**P<0.01vsmodel group.
图4AD模型组与FSD-C10治疗组小鼠小胶质细胞免疫组化染色和Westernblot检测结果的比较
讨 论
到目前为止,AD发病的机理仍然不清楚,主要学说包括胆碱能学说、β淀粉样蛋白瀑布学说、Tau蛋白假说、神经血管学说和氧化应激学说等,许多基因、细胞和分子的异常使得该病机制更为复杂,治疗更为困难,就目前对AD的认识程度,用于治疗AD并已获美国食品与药物管理局批准的药物包括胆碱酯酶抑制剂(多奈哌齐)、NMDA受体拮抗剂(美金刚)等[6]。这些药物只能缓解AD症状,但不能影响疾病的进展。
ROCK的异常激活可见于AD实验模型脑组织中,推测可能参与疾病的发生和发展。研究显示抑制ROCK可增加海马锥体神经元突触密度和长度,改善空间学习和记忆[7]。最近研究证实抑制ROCK可减少Aβ产生, 增加可溶性APP或单体Aβ[8-9]。因此,ROCK抑制剂成为AD防治的潜在药物靶点。我们的前期研究证实ROCK抑制剂Fasudil对AD小鼠具有较好的治疗效果,新型ROCK抑制剂FSD-C10,其效价与Fasudil等同,但对神经元毒性小、对血管影响小,安全窗相对较大。本研究试图观察FSD-C10干预对APP/PS1双转基因小鼠的治疗效果,并探讨可能的细胞分子机制。
Aβ沉积引起的老年斑及过磷酸化的Tau蛋白形成的神经原纤维缠结是AD的2个特征性病理改变[10]。BACE蛋白是调节Aβ生成的酶。其中Aβ是AD的核心致病物质,具有很强的自聚性,只要形成聚集体,就能表现出明显的神经毒性[11]。本研究结果显示FSD-C10干预明显提高APP/PS1小鼠的学习记忆能力,有效减少APP/PS1小鼠海马区AD的病理标志物Aβ沉积,抑制Tau蛋白磷酸化和BACE蛋白表达,说明FSD-C10具有成为治疗AD的潜能。Aβ在AD早期发挥启动致病的作用,一旦AD病程启动,Aβ在脑内特定区域聚集,使脑内的免疫细胞——小胶质细胞和星形胶质细胞活化并增殖,并过量释放NO、TNF-α、IL-6 等促炎因子,介导神经细胞的炎症损伤,触发免疫炎症级联反应[12]。大量炎症介质和炎症因子的释放使得中枢神经组织处在长期的炎症微环境中,可以造成神经突触损伤,线粒体功能障碍,神经元变性死亡[13]。已有多项研究认为胶质细胞介导的免疫炎症机制可能参与AD神经元变性的过程[14]。
TLRs/NF-κB信号通路激活所引起的炎性反应可能是AD发病中炎症损伤的一种重要机制。在AD病理进展中,Aβ沉积可引起TLR4表达增加,并且诱导NF-κB激活,从而促进多种炎症介质的表达和释放,进一步加重组织损伤[15-17]。本研究发现FSD-C10干预抑制TLRs/NF-κB炎症通路激活,有效改善APP/PS1小鼠脑组织的炎症微环境,从而减少对神经元损伤。
MG作为脑内常驻的免疫细胞,主要参与中枢神经系统的免疫和炎症反应,是 AD 炎症反应中最主要的炎症细胞,介导并贯穿AD 病理发展全程[18-19]。AD患者大脑中激活的小胶质细胞数量增加,特别是AD在Aβ沉积为老年斑的区域。应用PK11195 PET扫描也发现AD患者脑内呈现小胶质细胞激活成像。AD尸检也发现患者脑内伴有明显小胶质细胞反应,斑块周围有大量M1型小胶质细胞。激活的M1型小胶质细胞可分泌诸多炎症细胞因子和活性氧物质,能够引起神经元的凋亡[20]。小胶质细胞根据功能状态不同分为M1型和M2型,M1型可引起组织炎症性损伤,M2型具有抑制炎症反应和促进组织修复的作用。本实验发现FSD-C10干预抑制APP/PS1小鼠iNOS表达,而增加Arg-1表达,提示FSD-C10可以促进小胶质细胞M2极化,形成一个有助于抑制炎性和神经修复的微环境。
综上所述,FSD-C10有效改善APP/PS1双转基因小鼠的认知功能,其作用机制可能与调节小胶质细胞的极性,抑制炎症反应和改善炎症微环境有关。我们的研究仅为AD的治疗提供一个策略,有必要对FSD-C10治疗AD的机制进行深入研究。
[1] Carter MD, Simms GA, Weaver DF. The development of new therapeutics for Alzheimer’s disease[J]. Clin Pharmacol Ther, 2010, 88(4):475-486.
[2] Petratos S, Li QX, George AJ, et al. The β-amyloid protein of Alzheimer’s disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism[J]. Brain, 2008, 131(Pt 1):90-108.
[3] Hensel N, Rademacher S, Claus P. Chatting with the neighbors: crosstalk between Rho-kinase (ROCK) and other signaling pathways for treatment of neurological disorders[J]. Front Neurosci, 2015, 9:198.
[4] Krause DL, Müller N. Neuroinflammation, microglia and implications for anti-inflammatory treatment in Alzheimer’s disease[J]. Int J Alzheimers Dis, 2010, 2010:732806.
[5] Yu JZ, Li YH, Liu CY, et al. Multitarget therapeutic effect of Fasudil in APP/PS1 transgenic mice[J]. CNS Neurol Disord Drug Targets, 2017, 16(2):199-209.
[6] Neugroschl J, Sano M. Current treatment and recent clinical research in Alzheimer’s disease[J]. Mt Sinai J Med, 2010, 77(1):3-16.
[7] Couch BA, DeMarco GJ, Gourley SL, et al. Increased dendrite branching in AβPP/PS1 mice and elongation of dendrite arbors by fasudil administration[J]. J Alzheimers Dis, 2010, 20(4):1003 -1008.
[8] Lane RF, Gatson JW, Small SA, et al. Protein kinase C and rho activated coiled coil protein kinase 2 (ROCK2) modulate Alzheimer’s APP metabolism and phorylation of the Vps10-domain protein, SorL1[J]. Mol Neurodegener, 2010, 5:62.
[9] Ma QL, Yang F, Frautschy SA, et al. PAK in Alzheimer disease, Huntington disease and X-linked mental retardation[J]. Cell Logist, 2012, 2(2):117-125.
[10] 王明华, 周柏玉, 付雪锋. β淀粉样蛋白在老年痴呆症突触活动改变中的研究进展[J]. 神经解剖学杂志, 2015, 31(5):655-658.
[11] Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies[J]. Cell, 2012, 148(6):1204-1222.
[12] 刘绪华, 王孝庆, 王中苏, 等.姜黄素减弱 Aβ25-35致大鼠原代小胶质细胞的神经炎症反应[J]. 中国病理生理杂志, 2016, 32(9):1635-1641.
[13] Zhang F, Jiang L. Neuroinflammation in Alzheimer’s di-sease[J]. Neuropsychiatr Dis Treat, 2015, 11:243-256.
[14] Glass CK, Saijo K, Winner B, et al. Mechanisms underlying inflammation in neurodegeneration[J]. Cell, 2010, 140(6):918-934.
[15] Combs CK, Karlo JC, Kao SC, et al. β-Amyloid stimulation of microglia and monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis[J]. J Neurosci, 2001, 21(4):1179-1188.
[16] Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer’s disease, role of cytokines[J]. Sci World J, 2012, 2012:756357.
[17] Heneka MT, O’Banion MK. Inflammatory processes in Alzheimer’s disease[J]. J Neuroimmunol, 2007, 184(1):69-91.
[18] Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease[J]. Nat Rev Immunol, 2011,11(11):775-787.
[19] 聂 盼, 刘 勇, 杨学森, 等. 小胶质细胞体外吞噬模型的建立及相关炎症因子的研究[J]. 中国病理生理杂志, 2011, 27(9):1848-1851.
[20] Simi A, Tsakiri N, Wang P, et al. Interleukin-1 and inflammatory neurodegeneration[J]. Biochem Soc Trans, 2007, 35(Pt 5):1122-1126.
(责任编辑: 林白霜, 罗 森)
突触后突触结合蛋白在长时程增强过程中介导AMPA受体的胞吐作用
N-甲基-D-天冬氨酸(N-methyl-D-aspartate, NMDA)受体依赖性长时程增强(long-term potentiation, LTP)引起的突触联系增强具有重塑神经环路及调节学习和记忆的作用。在NMDA受体依赖性LTP诱导过程中,Ca2+内流会刺激突触α-氨基-3-羟基-5-甲基-4-异噁唑丙酸(α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid, AMPA)受体的募集,从而强化突触联系。然而,Ca2+诱导AMPK受体募集的机制尚未明确。Wu等的研究发现,在小鼠海马CA1区的锥体神经元中同时阻断突触结合蛋白1(synaptotagmin-1, Syt1)和突触结合蛋白7(synaptotagmin-7, Syt7)的突触后表达可拮抗LTP,但单独阻断其中一个蛋白则无此作用。野生型Syt7的表达可恢复LTP,而Ca2+结合缺失突变型Syt7则不会有这种作用。阻断Syt1和Syt7的突触后表达不会阻碍突触的基础传递,不减少突触或突触外的AMPA受体水平,也不会改变其他AMPA受体转运活动。此外,负显性突变Syt1(抑制Ca2+依赖性突触前囊泡的胞吐作用)的表达同样会阻断Ca2+依赖性突触后AMPA受体的胞吐作用,从而拮抗LTP。该研究结果表明,突触后Syt1和Syt7在LTP过程的AMPA受体Ca2+依赖性胞吐作用中是额外的Ca2+感受器,从而阐释了介导LTP的AMPA受体募集的一个简单机制。
Neuron, 2017, 544(7650):316-321(李 伟)
EffectofFSD-C10onmodulationofinflammatorymicroenvironmentinanAlzheimerdiseasedoubletransgenicmousemodel
GU Qing-fang1, YU Jie-zhong1, WU Hao1, LI Yan-hua1, FAN Hui-jie2, CHAI Zhi2, WANG Qing2, XIAO Bao-guo1, 3, MA Cun-gen1, 2
(1InstituteofBrainScience,DatongUniversity,Datong037009,China;2"2011"CollaborativeInnovationCenter/ResearchCenterofNeurobiology,ShanxiUniversityofTraditionalChineseMedicine,Taiyuan030024,China;3InstituteofNeurology,HuashanHospital,FudanUniversity,Shanghai200025,China.E-mail:macungen2001@163.com)
AIM: To explore the therapeutic effect of a novel Rho kinase inhibitor FSD-C10 on β-amyloid protein precursor (APP)/presenilin-1 (PS1) double transgenic mice.METHODSThe transgenic mice overexpressing humanAPPwith the Swedish mutation (695) and humanPS1 with ΔE9 mutation at the age of 8 months were used in this study. The mice were randomly divided into model group and FSD-C10 intervention group, and wild-type mice at the same age served as normal controls. The mice in FSD-C10 intervention group were treated with FSD-C10 (25 mg·kg-1·d-1) for 2 months by intraperitoneal injection. The mice in model group and the wild-type mice were injected with saline in the similar manner. Morris water maze (MWM) test was applied to examine the capacity of learning and memory. The Aβ1-42deposition, Tau protein phosphorylation, and the expression of β-site APP-cleaving enzyme (BACE) as well as inflammatory molecules, such as TLR-4 and NF-κB, and M1/M2 microglial markers, such as iNOS and Arg-1, were determined by the methods of immunohistochemistry and Western blot.RESULTSCompared with model group, FSD-C10 significantly improved the learning and memory abilities ofAPP/PS1 double transgenic mice, accompanied by reduced Aβ1-42deposition, Tau protein phosphorylation and BACE expression in the hippocampus. The intervention of FSD-C10 decreased the protein levels of TLR-4 and p-NF-κB, reduced the expression of iNOS and increased the expression of Arg-1 in the brain tissues.CONCLUSIONThe novel Rho kinase inhibitor FSD-C10 improves the capacity of spatial learning and memory inAPP/PS1 double transgenic mice, which may be related to the inhibition of TLRs/NF-κB signaling pathway, the reduction of the secretion of inflammatory molecules and the polarization of anti-inflammatory M2 microglia, thus improving the inflammatory microenvironment of the brain inAPP/PS1 double transgenic mice.
APP/PS1 double transgenic mice; FSD-C10; Inflammatory microenvironment; TLRs/NF-κB pathway
R363; R741
A
10.3969/j.issn.1000- 4718.2017.10.001
1000- 4718(2017)10- 1729- 09
2017- 03- 27
2017- 07- 20
国家自然科学基金资助项目(No. 81471412; No. 81272163);山西省国际科技合作项目(No. 2013081058);山西中医学院“2011”培育计划项目(No. 2011PY-1);大同市科技局基础研究计划项目(No. 2017136; No. 2014105-1);大同大学校科研项目(No. 2016K10)
△通讯作者 Tel: 0351-3179809; E-mail: macungen2001@163.com
▲并列第1作者
杂志网址: http://www.cjpp.net
