锂辉石表面环烷酸吸附行为的量子化学
2017-10-13何桂春张慧婷华亚南
何桂春,张慧婷,华亚南,蒋 巍
锂辉石表面环烷酸吸附行为的量子化学
何桂春1,张慧婷1,华亚南1,蒋 巍2
(1. 江西理工大学资源与环境工程学院,赣州 341000;2. 中南大学资源加工与生物工程学院,长沙 410000)
采用量子化学计算、红外光谱分析研究了环烷酸在锂辉石表面的吸附行为。通过计算表面能选取锂辉石(110)面为最佳解理面,并构建了无虚频的稳定环烷酸分子模型,运用分子动力学模拟得到环烷酸与锂辉石(110)面的最佳吸附构型。对最优吸附构型进行了量子化学计算,研究了其结构、电荷分布、差分电荷密度以及态密度等。结果表明:锂辉石(110)面吸附环烷酸分子后表面原子都向锂辉石内部弛豫,环烷酸吸附后的构型发生明显的变化,其羰基氧原子负电性得到增强,非羰基原子的负电性得到削弱,环烷酸与锂辉石的作用是通过羰基氧原子实现的。环烷酸与锂辉石吸附后,锂辉石(110)面的电子云重新分配,氧原子的电荷云密度增强并向周围区域发散开来,环烷酸整体态密度峰左移且费米能级附近的态密度峰由价带变为导带,体系趋向于稳定存在。红外光谱分析进一步表明:环烷酸在锂辉石表面吸附是物理吸附作用。
量子化学计算;锂辉石(110)面;环烷酸;吸附构型
目前锂辉石的选别方法以浮选法研究最多[1]。在锂矿物浮选药剂及其基础理论方面国内外学者有了一定的研究。MOON等[2]从锂辉石的表面晶体化学特性出发,研究了油酸盐−锂辉石间相互作用关系。PRADIP[3]的研究表明矿浆中的一些溶盐离子(Ca2+、Fe3+、Mg2+等)不仅活化锂辉石,同时也活化脉石矿物,使锂辉石浮游性下降。王绍艳等[4]研究了环烷酸对萤石和重晶石的捕收作用,研究结果表明,环烷酸对萤石和重晶石的捕收作用具有较好的选择性,在弱酸性至中性介质中,环烷酸对萤石和重晶石捕收能力的选择性比油酸强。葛英勇等[5]通过红外光谱分析研究了环烷酸浮选萤石、赤铁矿的作用机理,研究结果表明:环烷酸通过化学吸附和物理吸附在这两种矿物的表面,由此导致其疏水可浮。
目前采用量子化学研究矿物晶体性质多集中于对硫化矿的计算。陈建华等[6−11]用量子化学的方法研究了方铅矿、黄铁矿、闪锌矿等硫化矿浮选行为。YEKELER等[12]采用密度泛函的方法研究了一组含硫捕收剂与银离子的相互作用,用捕收剂最高占据轨道能量及与银离子相互作用能大小判断捕收剂的捕收能力,计算结果与实验一致。孙伟等[13]利用密度泛函方法计算黄铜矿捕收剂的前线轨道的性质。采用量子化学方法计算氧化矿性质的研究较少。何桂春等[14]基于密度泛函理论研究了锂辉石晶体的能带结构、态密度、Mullinken布居以及前线轨道与锂辉石可浮性之间的关系。彭兰等[15]通过量化计算分析了胺类阳离子捕收剂各物化参数对实际捕收能力的影响,结果表明胺类阳离子捕收剂对铝硅酸盐矿物捕收性能的决定因素在于极性基团。
关于捕收剂与矿物的作用机理大多是通过红外光谱、等温吸附曲线、Zeta电位等方法进行研究得到,而捕收剂与矿物作用机理通过量子化学计算研究却鲜有报道。本文作者通过对锂辉石表面吸附环烷酸分子的动力学模拟得到的最优吸附构型的量子化学计算,得到锂辉石与环烷酸之间相互作用的构效关系,从而对环烷酸起捕收作用的机理进行研究。
1 实验
1.1 模型的选取
1.1.1 锂辉石表面的选取
锂辉石晶体结构来自于文献[14]中的晶体结构,如图1(a)所示。由图1(a)可知,锂辉石断裂面主要由Al—O键和Si—O键组成,皆属于高能键。其中存在着两种Al—O键和Si—O键。当Al原子与配位数为2的O原子的键长为1.83 nm,与配位数为3的O原子的键长为2.03 nm;Si原子与配位数为2的O原子的键长为1.59 nm,与配位数为3的O原子的键长为1.65 nm。由于Al—O键的键长要比Si—O键的键长长,Al—O键比Si—O键容易断裂,所以在锂辉石形成的表面中,主要以断裂的Al—O键为主。(110)表面、(010)表面和(100)表面为锂辉石的主要晶面[16]。
通过计算{110}、{010}和{100}面的表面能,可以确定锂辉石的最主要解理面。由Material Studio建立的Slab表面模型实际仍然具有三维的超晶胞结构。每个表面模型具有上下两个表面,表面能计算公式[17]为
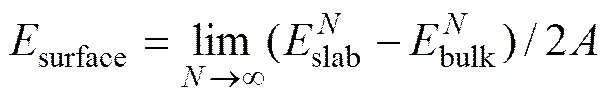
图1 锂辉石晶体模型及经COMPASS力场处理后的(110)面[14]
Fig. 1 Model of spodumene crystal(a) and (110)surface(b) treated by COMPASS field[14]
1.1.2 环烷酸分子模型的建立
在锂辉石(110)面进行吸附药剂的分子动力学模拟之前要构建药剂的分子结构模型。利用Material Studio中的Visualizer模块绘制出环烷酸的分子模型,并利用Dmol3对药剂分子进行结构优化并对结构优化后的模型进行声子谱计算;如果环烷酸分子有虚频的出现,则继续进行结构优化,直至找到无虚频的稳定结构。对Dmol3的设置参数见表1,优化后的环烷酸模型如图2(a)所示,环烷酸分子的声子谱见图2(b)。由图2(b)可知,优化后的环烷酸分子声子谱图中为负的横坐标没有值的出现,说明结构不存在虚频,所构建优化的环烷酸分子为合理稳定结构的分子,该结构可以用于后面的分子模拟和量子化学计算。
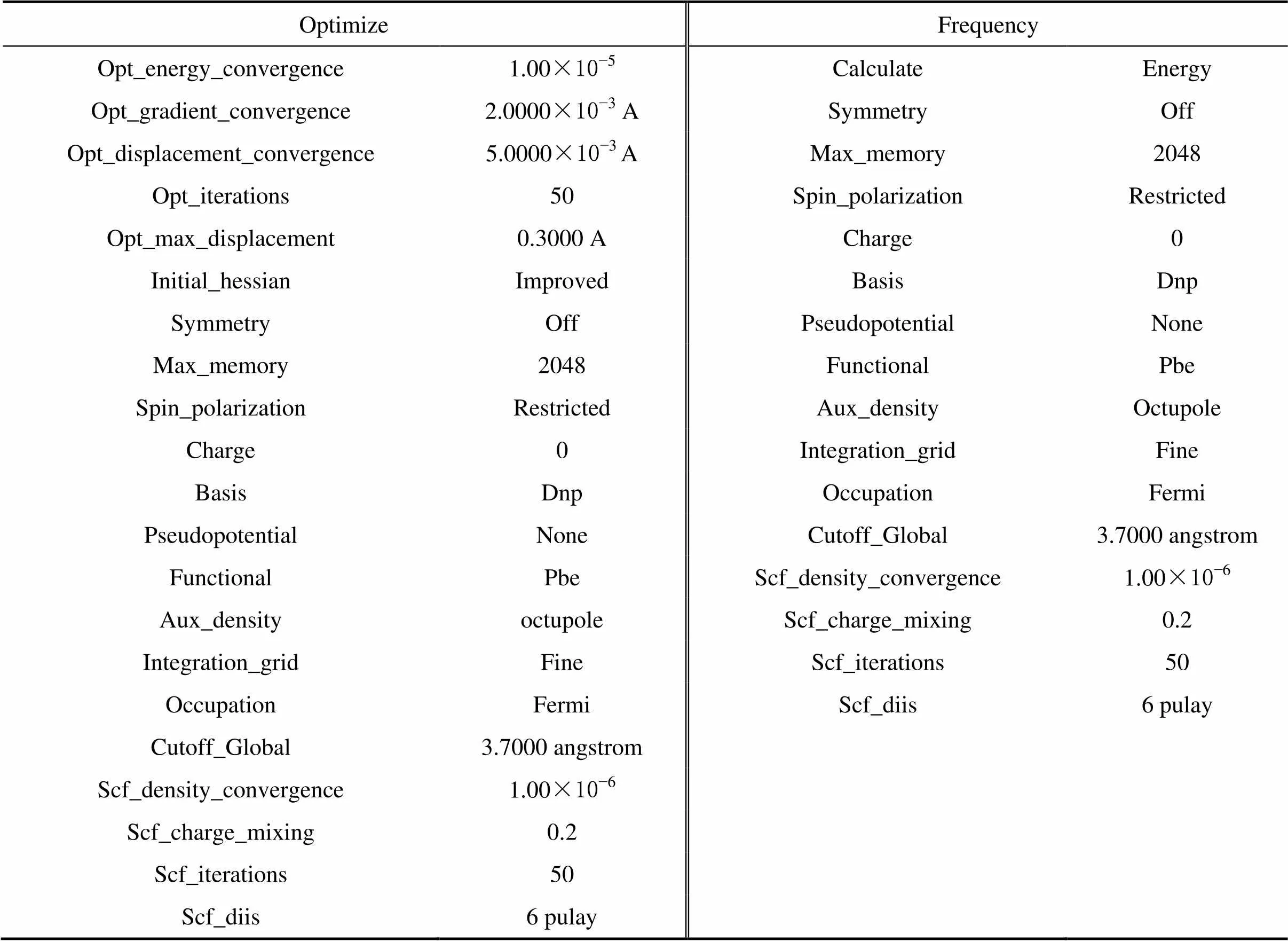
表1 Dmol3参数设置表

图2 环烷酸分子优化构型及其声子谱图
1.1.3 锂辉石(110)面吸附环烷酸分子的动力学模拟
将结构优化后的环烷酸放置在经过优化的锂辉石(110)表面上,如图3(a)所示。由于锂辉石(110)面与环烷酸产生作用的为表面的几层原子,因此在模拟的时候,对锂辉石表面下的原子实行了固定,只允许表面的那层原子发生弛豫。由于构型是周期性边界条件下的,为了防止重复的力场的干扰,在环烷酸与锂辉石(110)面初始构型中设置了6 nm的真空层。经过结构优化后的结构如图3(b)所示。从图3(a)和(b)可知,经过优化后的环烷酸分子发生了变化,相对于初始放置的构型,环烷酸分子中的羰基氧原子几乎垂直于锂辉石(110)表面,是环烷酸分子中最靠近锂辉石(110)面的原子。然而,这只是一个可能的优化构型,为了探寻环烷酸吸附在锂辉石(110)面的最优构型,应该尽可能地选取多的构型作比较。因此,应用Material Studio中的Forcite模块中的Quench方法,模拟过程中采用正则系综(NVT),控温采用Nose方法控制为298K,时间步长为1 fs,动力模拟时间为100 ps,采用修正的Ewald加合方法计算静电力作用,范德华作用力的截断半径为15.5 Å,采用Smart最小化法优化几何构型,收敛精度为fine。据在100 ps时间的分子动力学模拟的过程中,每隔5000步选择一个构型,总共选取21个构型进行能量计算。最后选取能量最低的一个构型即为锂辉石(110)面吸附环烷酸的最优构型。图4所示为100 ps时间内锂辉石(110)面吸附环烷酸的21个结构的能量图。从图4可知,第2至第4个构型为最低能量构型,如图3(c)所示。从图3(c)可知,相对于优化构型(见图3(b)),环烷酸分子以羰基氧原子为基点近乎垂直于穴位的作用于锂辉石(110)面,其中环烷酸分子中的羰基氧原子距锂辉石(110)面中的Li原子距离为0.1907 nm。
由锂辉石(110)面吸附环烷酸分子的最优构型计算出锂辉石(110)面与环烷酸分子的相互作用能,为−1282.92 kJ/mol,为较大的负值,说明在锂辉石(110)面对环烷酸分子具有较大的吸附能力。
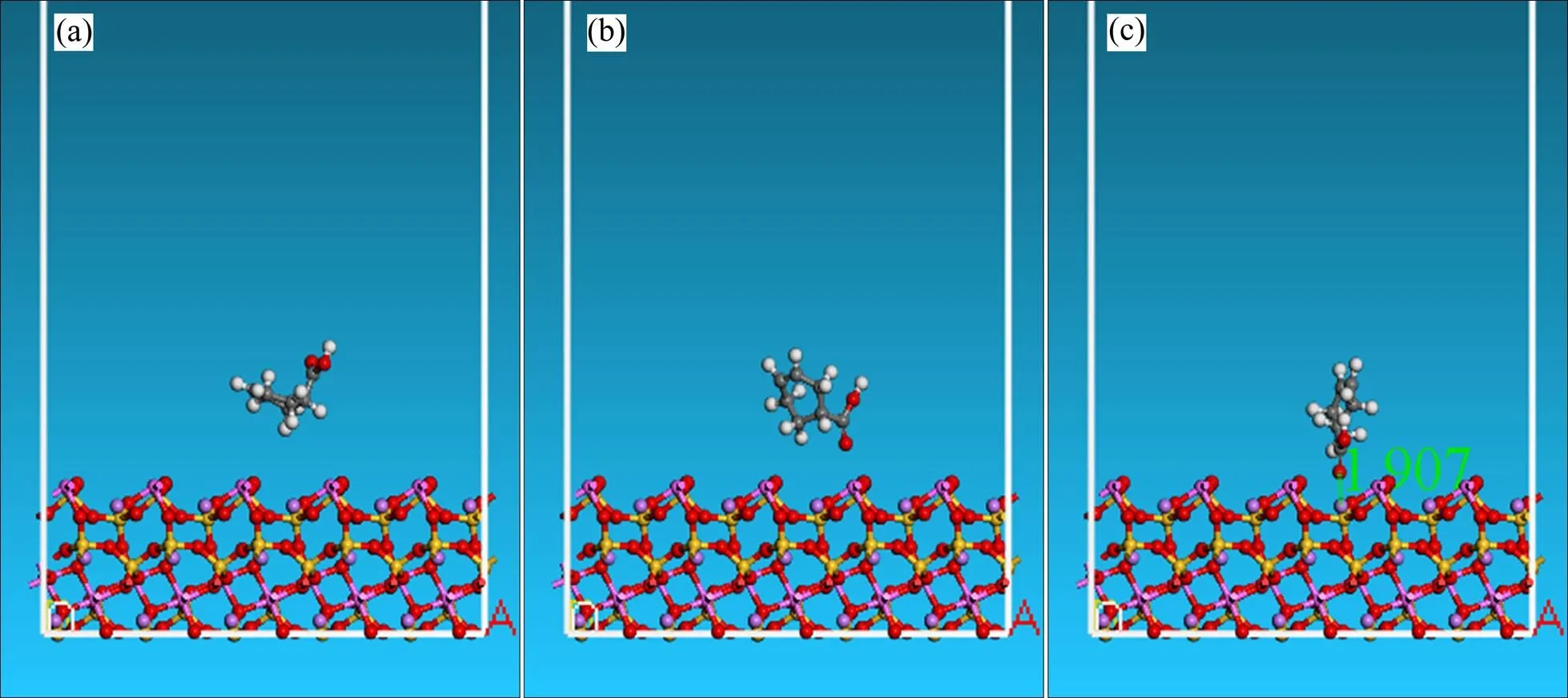
图3 真空环境下锂辉石(110)面与环烷酸络合构型
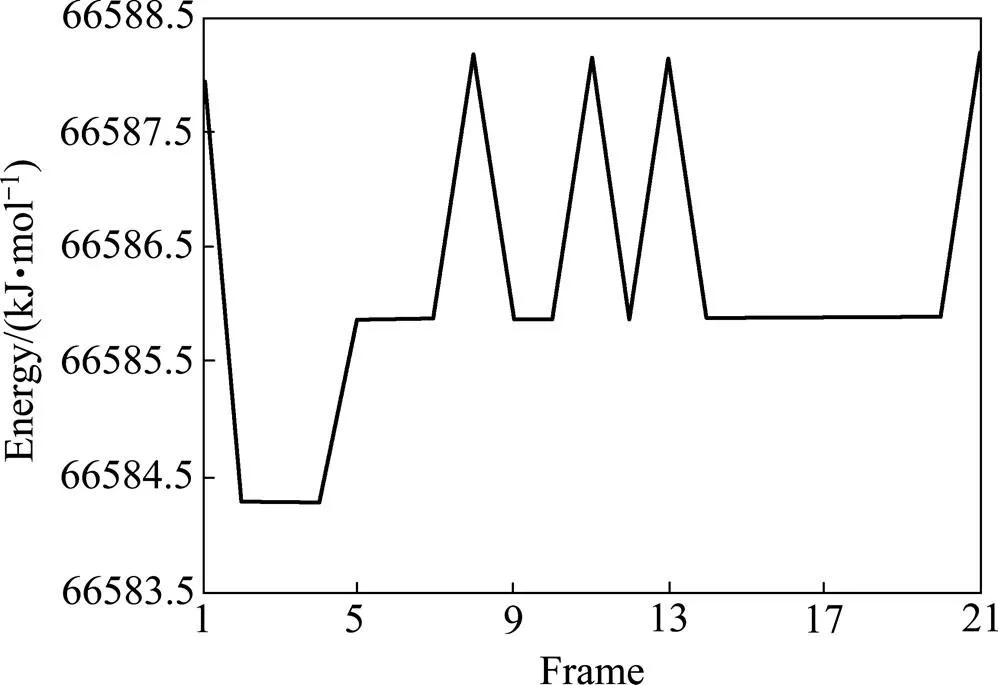
图4 100 ps内锂辉石(110)面吸附环烷酸各构型的能量图
1.2 计算方法
在material studio软件中的Castep[18−21]模块中基于密度泛函理论[22−23]对所得到的锂辉石吸附环烷酸最优构型进行量子化学计算,从计算结果中对其进行结构分析、电荷分析、差分电荷密度分析以及态密度分析研究环烷酸与锂辉石的相互作用机理。本研究中对最优构型进行量子化学性质计算的条件设为:采用 GGA 下的 PW91 交换关联泛函;采用 300 eV 平面波截断能和 2×2×1 的 Monkhorst-Pack k 点取样密度;采用超软赝势来描述价电子和核的相互作用。几何优化的收敛标准为原子位移的收敛阈值为0.0002 nm,原子间作用力的收敛阈值为 0.8 eV/nm,原子间的内应力收敛阈值为 0.1 GPa,最大能量改变的收敛阈值为2.0×10−5eV/atom;自洽迭代收敛精度为 2.0×10−6eV/atom;原子的赝势计算选取的价电子分别为H 1s1;Li 1s22s1;C 2s22p2;O 2s22p4;Al 3s23p1;Si 3s23p2。
2 结果与讨论
2.1 吸附环烷酸分子的锂辉石(110)表面结构弛豫
在保持吸附构型吸附位点附近原子完整且不影响吸附构型的条件下将最优构型进行简化,这有利于更高效的完成量子化学计算,简化后的构型如图5所示。表2列出了吸附环烷酸后锂辉石(110)面的弛豫情况,正号代表向锂辉石外部弛豫,负号代表向锂辉石内部弛豫。由表2可知,锂辉石表面由于吸附环烷酸的作用,都发生了弛豫,而且都向锂辉石表面的内部弛豫。由各原子的位移距离可知,位于环烷酸吸附位点的1、2、3、4、5、6号原子向内部弛豫的程度要比与吸附位点同样结构的12、13、14、15、16、17号原子大,说明环烷酸吸附位点附近对锂辉石表面弛豫的影响要大于锂辉石表面其他地方。
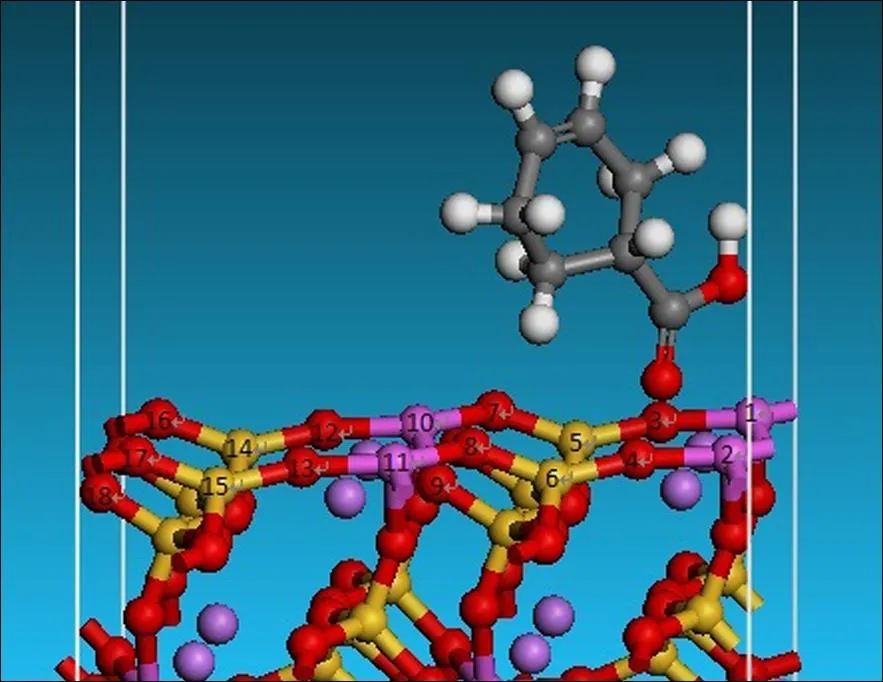
图5 锂辉石(110)面吸附环烷酸的简化构型

表2 锂辉石(110)表面原子配位及位移
2.2 表面原子电荷分析
由表3可知,吸附环烷酸后锂辉石表面一部分原子所带净电荷的值发生了变化,净电荷值发生变化的原子为1 (Al)、2 (O)、8(O)、9(Al)、10(Li)、12(Al)、16(Si)、20(Al)、21(O),除了16(Si)外,这些原子刚好位于锂辉石(110)表面吸附环烷酸的吸附位点附近;而离吸附位点较远的原子如13(O)、14(Si)、15(O)、17(O)、18(Si)、19(O)净电荷值并没有发生变化,吸附位点附近的Si原子3、5、7所带净电荷也没有发生变化。净电荷发生变化的原子中1(Al)、9(Al)、10(Li)在发生吸附作用后所带净电荷增加,由图6可知,这3个原子为锂辉石(110)表面与环烷酸直接发生作用的3个金属原子。说明当锂辉石(110)表面吸附环烷酸时,锂辉石(110)表面吸附环烷酸位点附近的原子所带净电荷会发生变化,而且所带净电荷增加的原子为与环烷酸直接作用的金属原子。
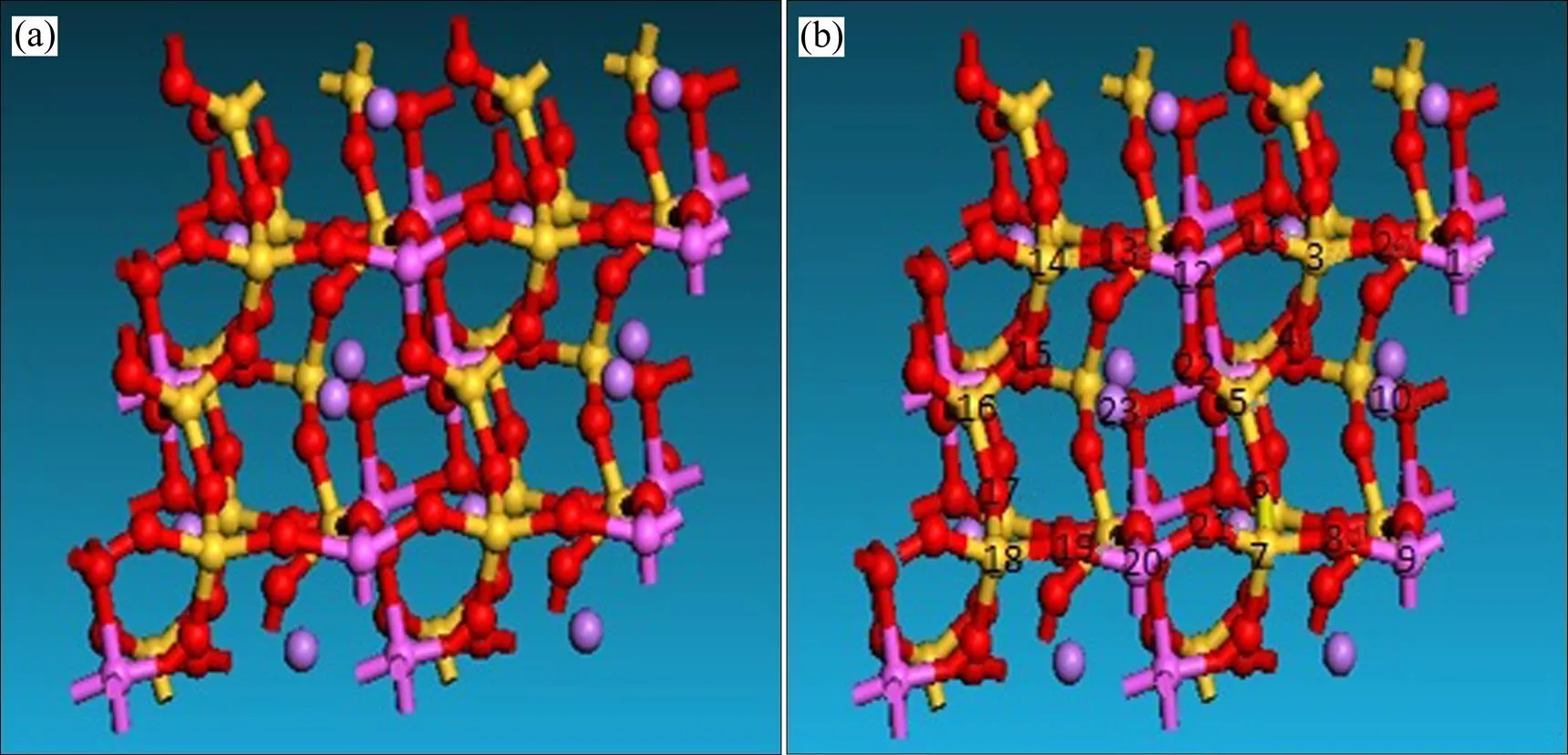
图6 吸附环烷酸前后的锂辉石(110)面
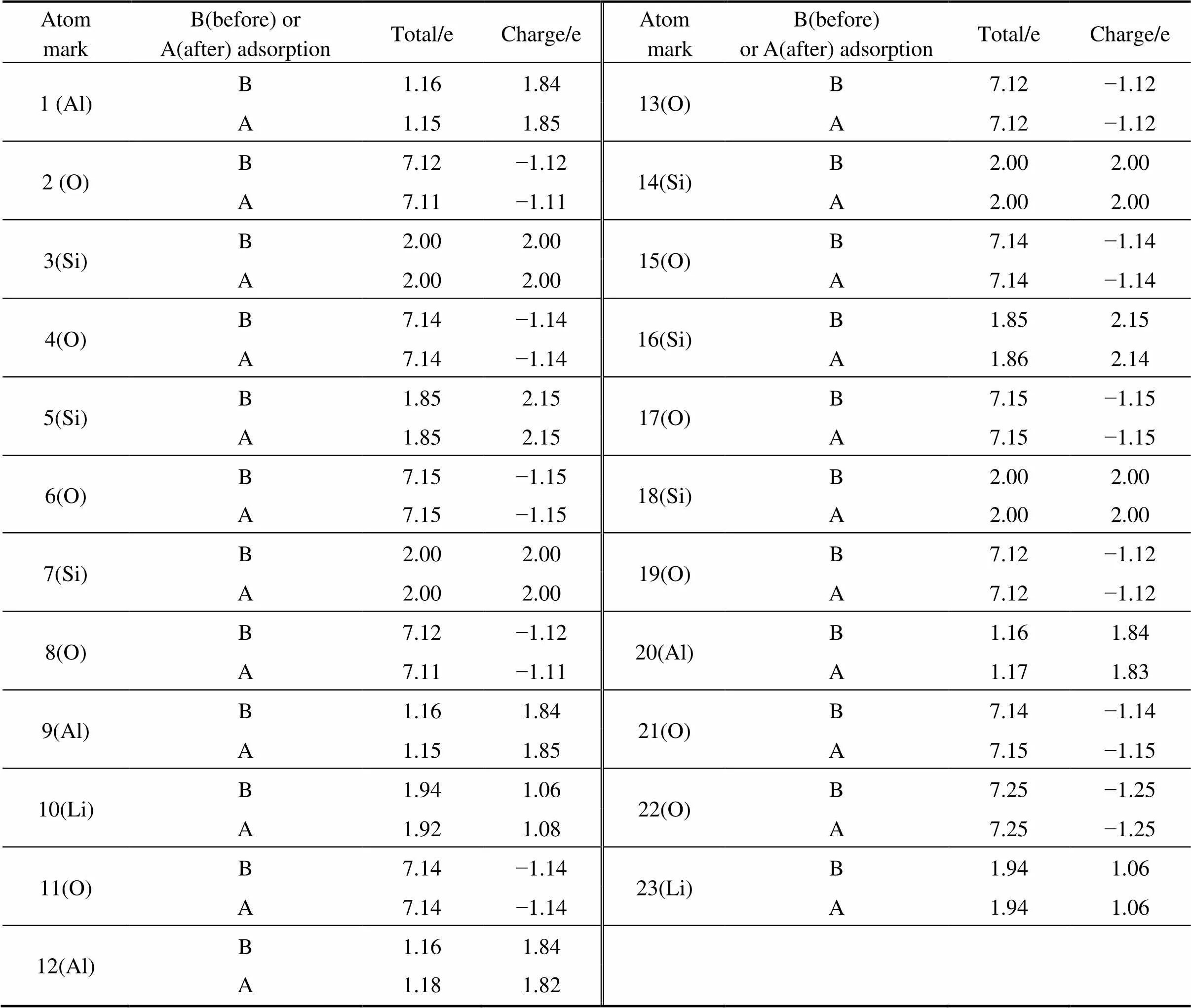
表3 吸附环烷酸前后的锂辉石(110)表面的Mulliken电荷布居
2.3 吸附前后的环烷酸分子电荷分析
图7所示为锂辉石(110)表面发生吸附前后环烷酸的构型图。由图7可以看到,环烷酸在锂辉石(110)表面发生吸附前后的构型发生了明显的变化。由表4吸附前后环烷酸分子的Mulliken电荷布居表可知,吸附改变了环烷酸分子内的H、C、O原子的净电荷,原子净电荷改变最明显的为环烷酸中的羧基中的原子1(H)、11(C)、18(O)、19(O)。环烷酸与锂辉石(110)面发生吸附作用的主要是羧基。当吸附发生后,羰基氧原子净电荷由−0.56e变为−0.57e,羰基氧原子的负电性得到增强;非羰基氧原子净电荷由−0.72e变为−0.56e,氧原子的负电性得到削弱。从图8也可以看到(蓝色区域表明电子缺失,红色区域表明电子富集),在环烷酸吸附位置处,也就是环烷酸与锂辉石(110)面发生相互作用的地方,电子云密度显著增强,氧原子电荷云密度增强并向周围区域发散开来,这些都是因为锂辉石(110)面吸附环烷酸后电子云重新分配的结果。

图7 在锂辉石(110)面发生吸附前后的环烷酸
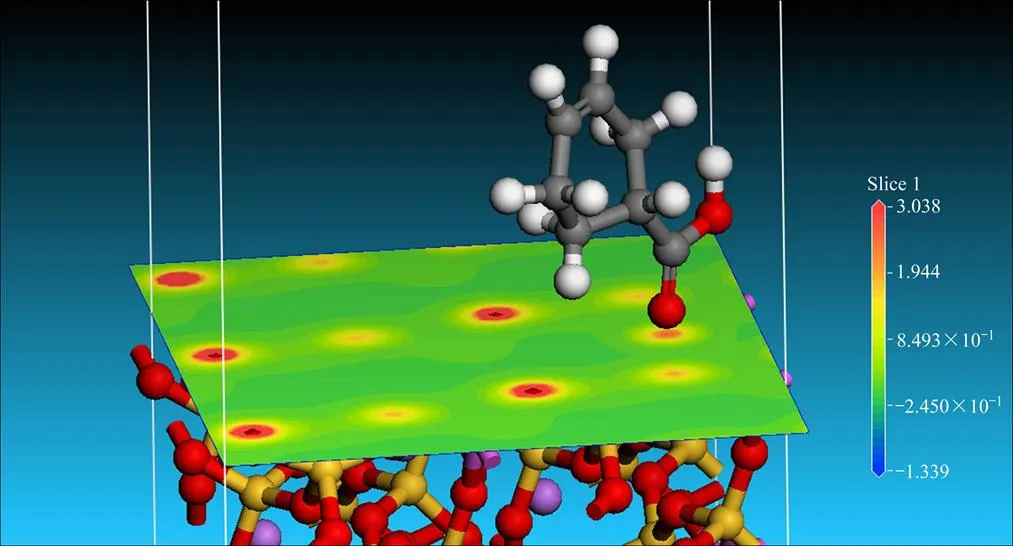
图8 锂辉石(110)表面吸附环烷酸分子的差分电荷密度
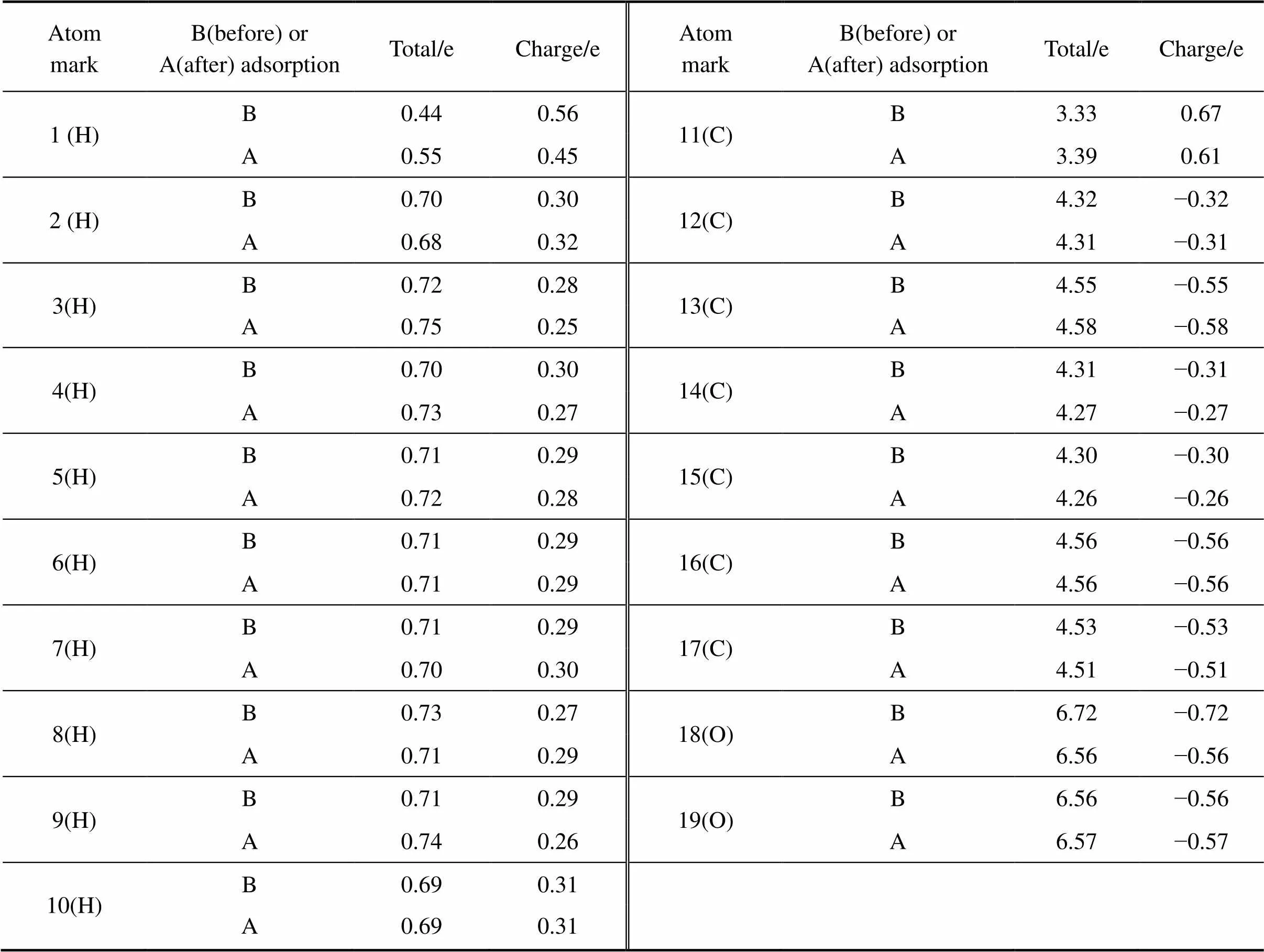
表4 在锂辉石(110)面发生吸附前后环烷酸分子的Mulliken电荷布居
2.4 环烷酸分子吸附对锂辉石(110)面态密度的影响
图9所示为环烷酸吸附前后锂辉石(110)表面O、Si原子的分态密度。吸附前后氧原子的s、p态密度变化不大,O的导带p态密度峰在环烷酸吸附后有所减弱。Si原子的态密度在环烷酸吸附表面后变化明显,在导带p态密度峰和s态密度峰显著减弱。
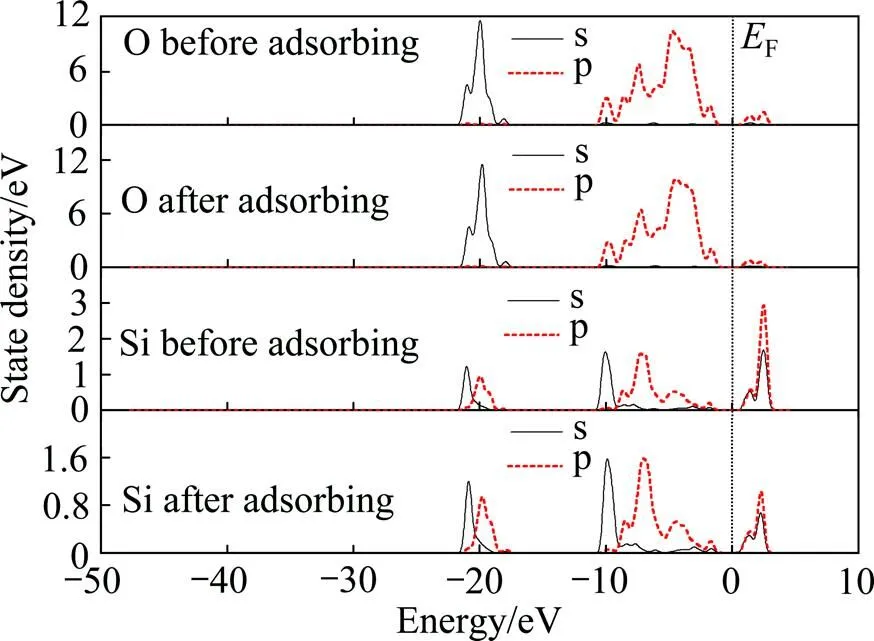
图9 环烷酸吸附前后锂辉石(110)表面O、Si态密度
图10所示为环烷酸吸附前后锂辉石(110)表面Al、Li原子的分态密度。由图10可知,Al原子态密度变化在吸附后主要在导带,p态密度和s态密度都由吸附前的两个峰变为一个峰,但是峰的位置更靠近费米能级的位置,说明活性得到增强。Li原子的态密度在价带变化明显,在−45 eV位置处由原先的一个接近8 eV的态密度峰变为两个4 eV左右的态密度峰。

图10 环烷酸吸附前后锂辉石(110)表面Al、Li态密度
图11所示为吸附前后环烷酸和锂辉石(110)表面态密度。由图11可见,吸附后环烷酸的态密度变化显著,整体态密度峰左移,费米能级附近的态密度峰由吸附前的价带变为导带,说明环烷酸吸附在锂辉石(110)面后发生了明显的变化;而锂辉石(110)面的态密度在吸附前后并无显著变化,且费米能级附近的s态密度峰值几乎没有变化。
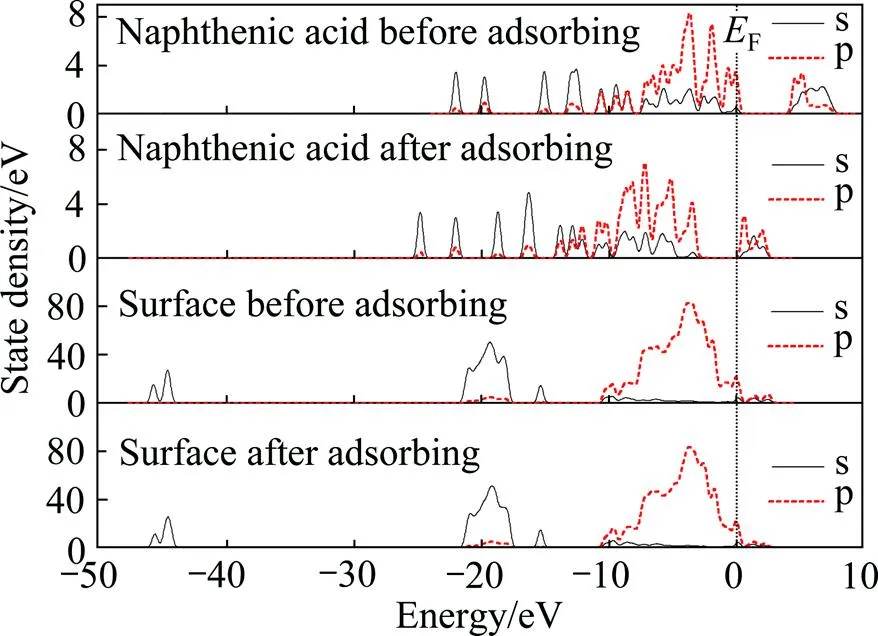
图11 吸附前后环烷酸和锂辉石(110)表面态密度
为了进一步验证环烷酸在锂辉石表现的吸附作用,对环烷酸、锂辉石以及环烷酸与锂辉石作用后的产物分别进行了红外光谱的检测(见图12)。
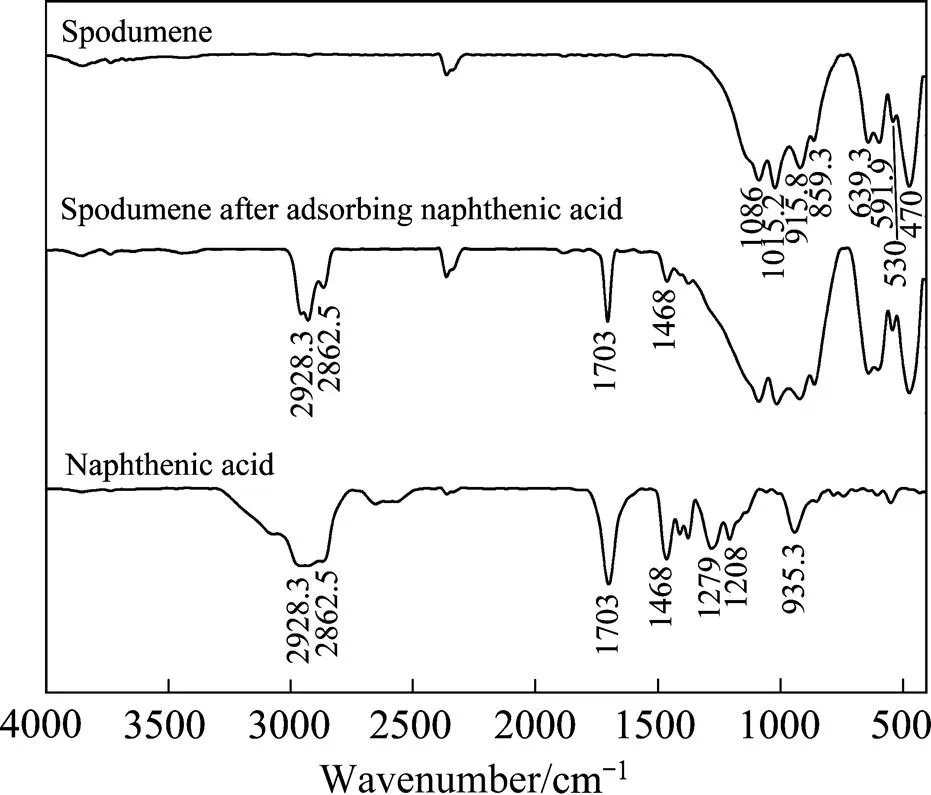
图12 锂辉石与环烷酸作用前后的红外光谱
2.5 环烷酸在锂辉石表面吸附产物的红外光谱分析
在锂辉石的红外光谱图中,1086、1015.2、915.8、859.3 cm−1处是Si—O伸缩振动吸收峰,639.3 cm−1处为Al—O伸缩振动吸收峰,591.9和530 cm−1处则为Al—O弯曲振动引起的,470 cm−1处为Li—O的振动吸收峰[24]。在环烷酸的红外光谱图中,2928.3和 2862.5 cm−1处是甲基亚甲基—C—H—键的伸缩振动共振峰,1703 cm−1处为—COO—中C=O的伸缩振动峰,1468 cm−1处为亚甲基剪式弯曲振动峰,1279、1208、935.3 cm−1处是—C—O—键的伸缩振动吸收峰[25]。
从红外图谱可以看出,锂辉石与环烷酸作用后,在2928.3、2862.5、1703、1468 cm−1波长处都有峰存在(见图12)。这些峰分别对应了环烷酸红外红外光谱图中的甲基亚甲基伸缩振动共振峰、羧酸根羰基的伸缩振动峰以及亚甲基剪式弯曲振动峰,表明环烷酸在锂辉石纯矿物表面有吸附作用。锂辉石与环烷酸作用后没有新的峰出现,表明这种吸附不是化学吸附而是物理吸附,这与理论计算结果相一致。
3 结论
1) 吸附环烷酸分子的锂辉石(110)面都向锂辉石的内部弛豫。
2) 环烷酸在锂辉石(110)表面发生吸附后的构型发生了明显的变化,吸附后的羰基氧原子净电荷由−0.56 e变为−0.57 e,羰基氧原子的负电性得到增强;非羰基氧原子净电荷由−0.72 e变为−0.56 e,氧原子的负电性得到削弱,环烷酸与锂辉石(110)表面的接触通过羰基氧原子实现。
3) 锂辉石(110)面吸附环烷酸后电子云重新分配,氧原子电荷云密度增强并向周围区域发散开来。吸附后环烷酸整体态密度峰左移,费米能级附近的态密度峰由吸附前的价带变为导带。锂辉石(110)面的态密度在吸附前后并无显著变化,且费米能级附近的s态密度峰值几乎没有变化。
4) 红外光谱分析结果表明,锂辉石与环烷酸作用后没有新的峰出现,环烷酸在锂辉石表面的吸附是物理吸附。结合量子化学计算结果和红外光谱分析结果可知,环烷酸分子通过羰基氧原子与锂辉石表面的金属原子发生了静电吸附作用从而吸附在锂辉石表面。
[1] 何桂春, 蒋 巍, 项华妹. 锂辉石选矿研究现状及展望[J]. 矿业研究与开发, 2014(4): 61−65. HE Gui-chun, JIANG Wei, XIANG Hua-mei. Research status and prospects of spodumene beneficiation[J]. Mining R&D, 2014(4): 61−65.
[2] MOON K S, DOUGLAS W F. Surface crystal chemistry in selective flotation of spodumene (LiAl[SiO3]2) from other aluminosilicates[J]. International Journal of Mineral Processing, 2003, 72: 11−24.
[3] PRADIP P. The science of reagents design: An historical perspective[J]. Transactions of the Indian Institute of Metals (India), 1997, 50(6): 481−494.
[4] 王绍艳, 李晓安, 薛问亚. 环烷酸对萤石和重晶石捕收作用的研究[J]. 非金属矿, 1996(6): 27−29. WANG Shao-yan, LI Xiao-an, XUE Wen-ya. Study of the collection effect of naphthenic acid on fluorite and barite[J]. Non-Metallic Mines, 1996(6): 27−29.
[5] 葛英勇.环烷酸浮选萤石、赤铁矿作用机理的红外光谱研究[J].非金属矿, 1991(3): 19−21. GE Ying-yong. Mechanism of naphthenic acid in the flotation of fluorite and hematite[J]. Non-Metallic Mines, 1991(3): 19−21.
[6] CHEN Jian-hua, CHEN Ye, LI Yu-qiong. Quantum-mechanical study of effect of lattice defects on surface nonproperties and copper activation of sphalerite surface[J]. Transactions of Nonferrous Metals Society of China, 2010, 20: 1121−1130.
[7] 李玉琼, 陈建华, 陈 晔, 郭 进. 黄铁矿(100)表面性质的密度泛函理论计算及其对浮选的影响[J]. 中国有色金属学报, 2011, 21(4): 919−926. LI Yu-qiong, CHEN Jian-hua, CHEN Ye, GUO Jin. Density functional theory calculation of surface properties of pyrite (100) with implications for flotation[J]. The Chinese Journal of Nonferrous Metals, 2011, 21(4): 919−926.
[8] 陈建华, 钟建莲, 李玉琼, 陈 晔, 郭 进. 黄铁矿、白铁矿和磁黄铁矿的电子结构及可浮性[J]. 中国有色金属学报, 2011, 21(7): 1719−1727. CHEN Jian-hua, ZHONG Jian-lian, LI Yu-qiong, CHEN Ye, GUO Jin. Electronic structures and floatability of pyrite, marcasite and pyrrhotite[J]. The Chinese Journal of Nonferrous Metals, 2011, 21(7): 1719−1727.
[9] 陈建华, 王 檑, 陈 晔, 李玉琼, 郭 进. 空位缺陷对方铅矿电子结构及浮选行为影响的密度泛函理论[J]. 中国有色金属学报, 2010, 20(9): 1815−1821. CHEN Jian-hua, WANG Lei, CHEN Ye, LI Yu-qiong, GUO Jin. Density functional theory of effects of vacancy defects on electronic structure and flotation of galena[J]. The Chinese Journal of Nonferrous Metals, 2010, 20(9): 1815−1821.
[10] 李玉琼, 陈建华, 陈 晔. 空位缺陷黄铁矿的电子结构及其浮选行为[J]. 物理化学学报, 2010, 26(5): 1435−1441. LI Yu-qiong, CHEN Jian-hua, CHEN Ye. Electronic structures and flotation behavior of pyrite containing vacancy defects[J]. Acta Phys-Chim Sin, 2010, 26(5): 1435−1441.
[11] 陈建华, 王进明, 龙贤灏, 郭 进. 硫化铜矿物电子结构的第一性原理研究[J]. 中南大学学报(自然科学版), 2011, 42(12): 3612−3617. CHEN Jian-hua, WANG Jin-ming, LONG Xian-hao, GUO Jin. First-principle theory on electronic structure of copper sulfides[J]. Journal of Central South University (Science and Technology), 2011, 42(12): 3612−3617.
[12] YEKELER H, YEKELER M. Reactivities of some thiol collectors and their interactions with Ag+ion by molecular modeling[J]. Applied Surface Science, 2004, 236(1/4): 435−443.
[13] 孙 伟, 杨 帆, 胡岳华, 何国勇, 刘文莉. 前线轨道在黄铜矿捕收剂开发中的应用[J]. 中国有色金属学报, 2009, 19(8): 1524−1532. SUN Wei, YANG Fan, HU Yue-hua, HE Guo-yong, LIU Wen-li. Application of frontier orbital in developing new collectors of chalcopyrite[J]. The Chinese Journal of Nonferrous Metals, 2009, 19(8): 1524−1532.
[14] HE G C, XIANG H M, JIANG W, KANG Q, CHEN J H. First-principles theory on electronic structure and floatability of spodumene[J]. Rare Metals, 2014, 33(6): 742−748.
[15] 彭 兰, 曹学锋, 杜 平. 铝硅酸盐矿物捕收剂的设计研究[J].广西民族学院学报(自然科学版), 2005, 11(2): 90−93. PENG Lan, CAO Xue-feng, DU Ping. The studies of collectors to catch aluminum silicate minerals[J]. Journal of Guangxi University for Nationalities (Natural Science Edition), 2005, 11(2): 90−93.
[16] 周乐光. 矿石学基础[M]. 北京: 冶金工业出版社, 2007. ZHOU Le-guang. Foundation of ore[M]. Beijing: Metallurgical Industry Press, 2007.
[17] 钟建莲. 硫铁矿物电子结构及表面性质的第一性原理研究[D]. 南宁: 广西大学, 2011. ZHONG Jian-lian. First principle study on the electronic structure and surface property of sulfurous iron minerals[D]. Nanning: Guangxi University, 2011.
[18] XIE X D. Energy band theory of solids[M]. 1st ed. Shanghai: Fudan University Press, 2007.
[19] MARZARI N, VANDERBILT D, PAYNE M C. Ensemble density-functional theory for AB initio molecular dynamics of metals and finite-temperature insulators[J]. Phys Rev Lett, 1997, 79(7): 1337−1340.
[20] JONES R O, GUNNARSSON O. The density functional formalism, its applications and prospects[J]. Rev Mod Phys, 1989, 61(3): 689−746.
[21] KOHN W, SHAM L J. Self-consistent equations including exchange and correlation effects[J]. Phys Rev, 1965, 140(4A): A1133−A1138.
[22] CLARK S J, SEGALL M D, PICK C J, HASNIP P J, PROBERT M J, REFSON K, PAYNE M C. First principles methods CASTEP[J]. Zeitschrift fuer Kristallograhie, 2005, 220(5/6): 567−570.
[23] SEGALL M D, LINDAN P J D, PRONBERT M J, PICKARD C J, HASNIP P J, CLARK S J, PAYNE M C. First principles simulation: Idea, illustrations and the CASTEP code[J]. J Phys Cond Matt, 2002, 14(11): 2717−2744.
[24] 徐龙华, 董发勤, 巫侯琴, 王 振, 肖军辉. 油酸钠浮选锂辉石的作用机理研究[J]. 矿物学报, 2013, 33(2): 181−184. XU Long-hua, DONG Fa-qin, WU Hou-qin, WANG Zhen, XIAO Jun-hui. A Study on spodumene flotation mechanism by sodium oleate[J]. Acta Mineralogica Sinica, 2013, 33(2): 181−184.
[25] 陆婉珍, 袁洪富, 徐广通, 强冬梅.现代近红外光谱分析技术[M].北京: 中国石化出版社, 2000. LU Wan-zhen, YUAN Hong-fu, XU Guang-tong, QIANG Dong-mei. Modern near infrared spectroscopy analysis technology[M]. Beijing: China Petrochemical Press, 2000.
(编辑 何学锋)
Quantum chemistry of adsorption of naphthenic acid on surface of spodumene
HE Gui-chun1, ZHANG Hui-ting1, HUA Ya-nan1, JIANG Wei2
(1. Faculty of Resource and Environmental Engineering, Jiangxi University of Science and Technology, Ganzhou 341000, China;2. School of Resources Processing and Bioengineering, Central South University, Changsha 410083, China)
The mechanism of flotation of naphthenic acid on spodumene was studied by using the method of quantum chemistry calculation and infrared spectrum analysis. The paper has selected the (110) surface as the best cleavage plane by calculating different surfaces’ energy and has built stability naphthenic acid model with no imaginary frequency, then has got the best configuration for adsorption of naphthenic acid on spodumene (110) surface by using the method of molecular dynamics simulation. The structure, charge distribution, differential charge density and state density to the best adsorption configuration were studied. The results show that the atoms on the (110) surface relaxes to the spodumene inside when the naphthenic acid molecule is adsorbed on the (110) surface. At the same time, the configuration of the naphthenic acid changes obviously. The electronegative of the carbonyl oxygen atoms is stronger and those of the other oxygen atoms are weakened to the adsorbed naphthenic acid molecule. The interaction between naphthenic acid and spodumene is achieved by carbonyl atoms. After adsorption, the electron cloud is redistributed and the charge cloud density of the oxygen atoms increase and spread to the surrounding area on the spodumene (110) surface. The state density peak of the naphthenic acid molecule shifts to the left but the peak near the Fermi lever has changed from valence band to the conduction band. The adsorption structure tends to be stable. The infrared spectral analysis further shows that the naphthenic acid is physical adsorption on the spodumene surface.
quantum chemical calculation; spodumene (110) surface; naphthenic acid; adsorption configuration
Project(51104070) supported by the National Natural Science Foundation of China; Project (YC2013-S183) supported by the Graduate Student Special Funds of Jiangxi Province, China
2016-01-25; Accepted date:2016-06-23
JIANG Wei; Tel: +86-13177764834; E-mail: 799798300@qq.com
10.19476/j.ysxb.1004.0609.2017.04.021
1004-0609(2017)-04-0833-10
TD923
A
国家自然科学基金资助项目(51104070);江西省研究生创新专项资金项目(YC2013-S183)
2016-01-25;
2016-06-23
蒋 巍,博士;电话:13177764834;E-mail: 799798300@qq.com
