IFNGR1基因突变致分枝杆菌易感性疾病2例病例报告
2017-09-22应文静刘丹如孙金峤王文婕俞晔珩王晓川
应文静 刘丹如 孙金峤 王文婕 俞晔珩 王晓川
·论著·
IFNGR1基因突变致分枝杆菌易感性疾病2例病例报告
应文静 刘丹如 孙金峤 王文婕 俞晔珩 王晓川
目的 探讨IFNGR1基因突变致分枝杆菌易感性疾病(MSMD)的临床特征。方法 总结2例IFNGR1基因突变MSMD患儿的临床特征,ELISA方法检测干扰素-γ(IFN-γ)释放功能,流式细胞术检测IFNGR1蛋白表达,Sanger测序方法分析IFNGR1基因突变。结果 ①2例患儿均生后3月龄内出现卡介苗病,以卡介苗接种侧腋下淋巴结肿大为初始表现,并逐渐播散累及肺部、肠道、中枢和骨髓。确诊年龄分别为4岁和6岁。常规免疫功能(淋巴细胞亚群、免疫球蛋白、中性粒细胞呼吸爆发功能和补体)评估未见缺陷。②2例患儿的IFN-γ释放能力明显低下、IFNGR1蛋白表达均低于正常。③1例存在c.665 G>A(p.G219R)纯合突变,其父母均为c.665 G>A(p.G219R)杂合突变;1例存在c.665 G>A(p.G219R)和c.310 C>A(p.A104N)复合杂合突变,分别遗传自患儿母亲 [c.665 G>A(p.G219R)杂合突变]及父亲 [c.310 C>A(p.A104N)杂合突变]。其中1例患儿的突变为新发突变,既往无文献报道。④2例患儿在确诊前抗痨治疗效果不佳,确诊后加用IFN-γ,卡介苗感染得到控制,未见其他不良反应。结论IFNGR1基因突变可导致MSMD。卡介苗病患儿常规免疫评估无缺陷时,需考虑该病可能,相关蛋白检测、IFN-γ释放实验和基因分析有助于诊断。IFN-γ治疗有一定疗效。
分枝杆菌易感性疾病;IFNGR1基因; 卡介苗病
接种卡介苗是预防重症结核感染的主要手段。极少部分婴儿接种卡介苗后发生卡介苗病,宿主本身的免疫缺陷是导致卡介苗病的重要原因。分枝杆菌易感性疾病(MSMD)是一类对分枝杆菌易感的原发性免疫缺陷病,IFNGR1基因缺陷是其病因之一[1,2]。目前国内对该病的认识尚不足,现报告复旦大学附属儿科医院(我院)收治的2例IFNGR1基因突变致MSMD患儿的临床特征及相关实验室检查,以提高对该病的认识,促进早期诊断和治疗。
1 病例资料
例1 女,4岁,因“间断发热2月,右面部肿痛45 d”于2015年8月24日至我院就诊。
患儿足月顺产,生后患“湿肺”于当地医院治疗。1月龄(2011年7月)于左上臂接种卡介苗。图1显示外院治疗的经过,3月龄发现左腋下包块,呈进行性增大,伴发热和咳喘,抗生素治疗无效,淋巴结活检示抗酸染色++,诊断为“淋巴结核、肺结核、卡介苗病”,抗结核治疗18个月,肺部感染控制,淋巴结缩小。停药6个月后再次出现反复发热,精神差,频繁抽搐,在外院诊断为“结核性脑膜炎、脑积水”,口服异烟肼、利福平和乙胺丁醇,直至就诊我院。于我院就诊前2月患儿反复发热(37.5~38°C),午后和夜间明显,伴咳嗽、咳痰,1月余前出现右侧面部肿痛,伴局部皮温高,抗生素治疗未见好转,面部肿痛逐渐加重。
患儿父母身体健康,否认近亲结婚和遗传性疾病史。
体格检查:体温37.8℃,精神反应可。左侧腋下可见3 cm淋巴结活检术后瘢痕,无皮疹、出血点。面部肿胀,右侧明显;右侧颌下可及1个7 cm×6 cm肿块,左侧颌下可及1个2 cm×3 cm肿块,皮温高,质硬;颈部可及蚕豆到黄豆大淋巴结,质软,无压痛,余浅表淋巴结无肿大;扁桃体Ⅰ度肿大。双肺呼吸音粗,未及罗音。心音有力,律齐。腹软,肝脏肋下3 cm,剑突下4 cm,质软;脾肋下3 cm,质软;肠鸣音可。四肢肌力、肌张力可,神经系统查体阴性。
实验室检查:血常规WBC 16.1×109·L-1,N 0.71,Hb 133 g·L-1,PLT 269×109·L-1,CRP 60 mg·L-1,血沉38 mm·h-1,肝肾功能未见异常,血、尿、痰和脑脊液培养均阴性,呼吸道病毒、支原体、衣原体、EBV、CMV和T-SPOT阴性,PPD试验阳性,G试验阴性,自身抗体阴性,常规免疫功能(淋巴细胞亚群、Ig、补体和中性粒细胞呼吸爆发功能)评估未见异常(表1),骨髓细胞学检查结果未见异常。
影像学检查:胸部增强CT示,左侧腋下淋巴结肿大,左下肺门可疑小钙化灶。腹部增强CT示,肝脾肿大,脾脏多发低强化灶,后腹膜和腹腔多发淋巴结肿大伴钙化。头颅增强MR示,右侧额颞叶异常信号,脑积水。颌面部增强MR示,双侧颌面部及右侧口底软组织病变,双侧颈部多发小淋巴结,累及双侧下颌骨。全身骨扫描示,双侧下颌骨骨质病变。

例2 女,6岁,因“反复卡介苗感染6年余”于2017年1月6日至我院就诊。
患儿足月顺产,生后即于左上臂接种卡介苗。50日龄(2011年1月)时出现卡介苗接种部位红肿破溃。图1显示外院治疗经过,2月龄出现左腋下包块,进行性增大至4 cm×3 cm×4 cm,当地医院淋巴结穿刺抗酸染色++,考虑“淋巴结核”,抗结核药物治疗3个月后,淋巴结未见明显缩小,并出现发热、咳嗽、黏液便,于当地医院诊断为“淋巴结核、肺结核、肠结核、卡介苗病”,抗结核药物又治疗12个月,肺部感染控制,淋巴结缩小,遂停抗痨药物。停药12个月后再次出现反复发热,于外院诊断“非典型分枝杆菌感染、骨髓炎”,口服异烟肼、利福平、吡嗪酰胺和乙胺丁醇。
患儿父母身体健康,否认近亲结婚和遗传性疾病史。
体格检查:体温正常,精神反应可。左上臂卡疤2 cm×2 cm,左侧腋下可见2 cm淋巴结穿刺活检术,无皮疹、出血点。扁桃体Ⅰ度肿大;颈部可及数枚黄豆大小淋巴结,质中,无压痛,余浅表淋巴结无肿大。双肺呼吸音粗,未及罗音。心音有力,律齐。腹软,肝脏肋下2 cm,剑突下1 cm,质软;脾肋下未及;肠鸣音可。四肢肌力、肌张力可,神经系统查体阴性。
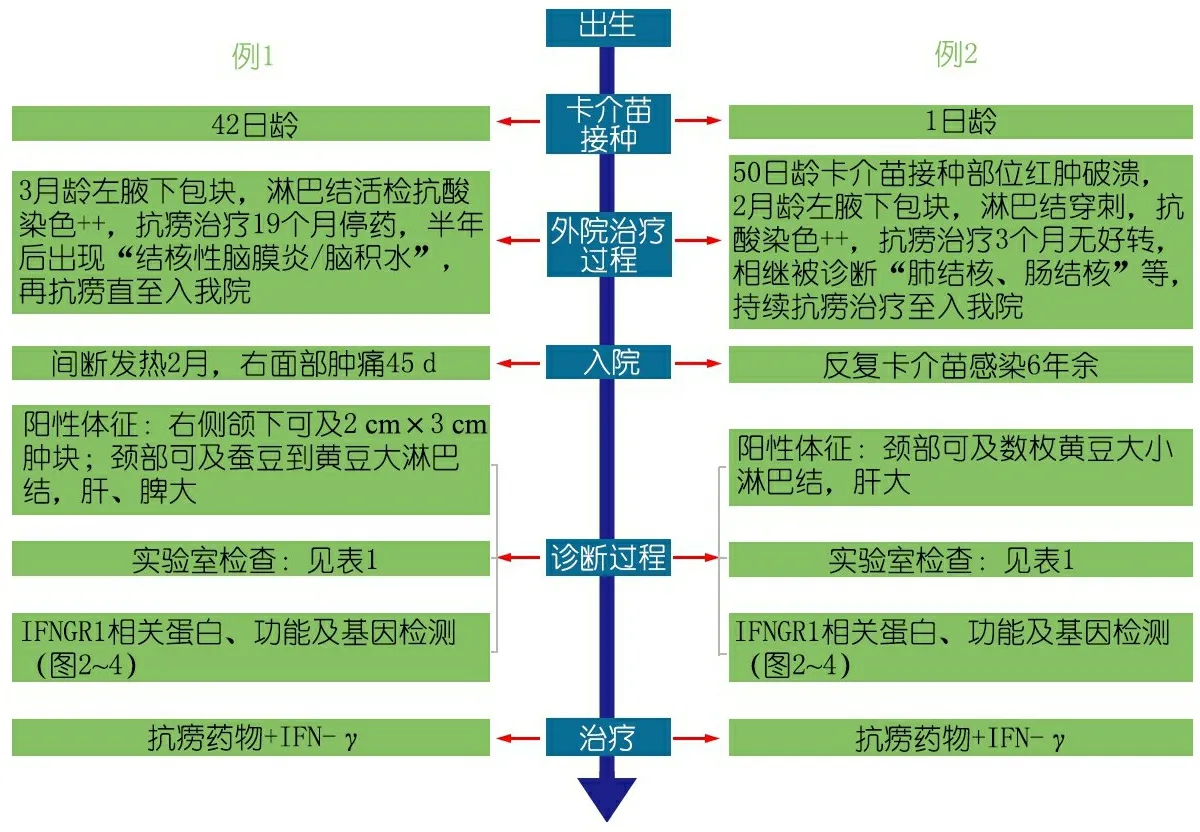
图1 患儿疾病病程重要信息时间轴
实验室检查:血常规WBC 13.42×109·L-1,N 0.53,Hb 98 g·L-1,PLT 413×109·L-1,CRP 94 mg·L-1,肝、肾功能未见异常,结核抗体阳性,PPD试验阳性,T-SPOT阴性,G试验阴性,常规免疫功能评估正常(表1)。
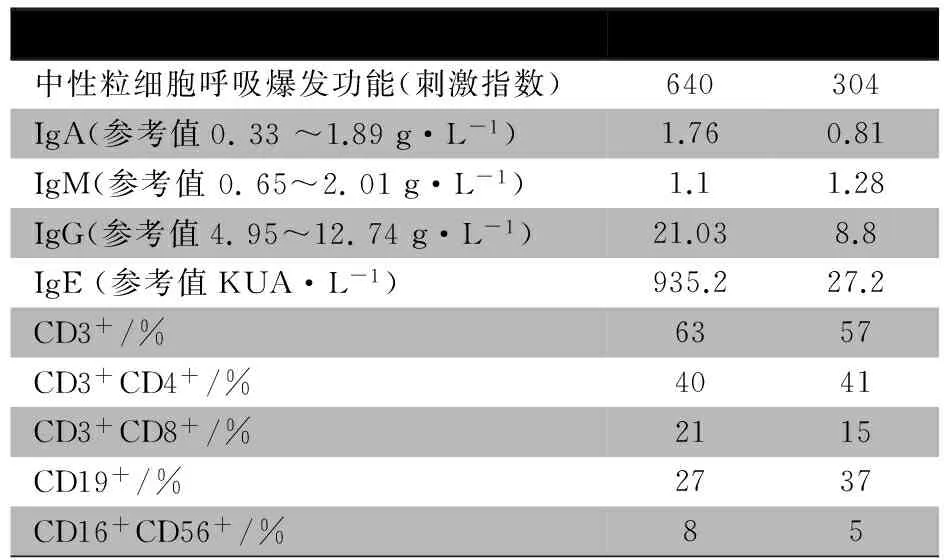
表1 2例患儿的常规免疫功能结果
影像学检查:胸部增强CT示,双肺炎症,左腋下多发钙化影。腹部增强CT示,肠道管壁增厚,结核可能,后腹膜及腹腔多发淋巴结肿大伴钙化。
诊断与治疗:予口服利福平、异烟肼和乙胺丁醇抗痨治疗。同时,行常规免疫功能检查,结果显示无缺陷,考虑患儿存在MSMD可能。取得患儿父母知情同意后,进行IFN-γ释放实验(ELISA法)、IFNGR1蛋白表达(流式细胞术检测)和基因检测(Sanger测序),结果提示IFN-γ产生能力明显低下(图2),IFNGR1蛋白不表达(图3),IFNGR1基因存在c.665 G>A(p.G219R)及c.310 C>A(p.A104N)复合杂合突变,患儿母亲为c.665 G>A(p.G219R)杂合突变,患儿父亲为c.310 C>A(p.A104N)杂合突变(图4)。IFNGR1基因突变所致MSMD诊断明确,在抗痨药物的基础上加用IFN-γ治疗,随访6个月,卡介苗感染控制良好,抗痨治疗中。
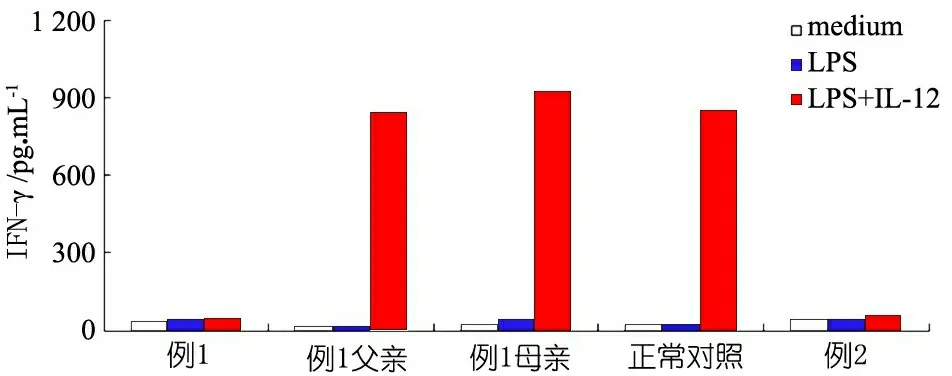
图2 IFN-γ产生能力测定(ELISA法)
注 2例患儿 IFN-γ产生能力均明显低下,给予LPS或LPS+IL-12刺激无反应
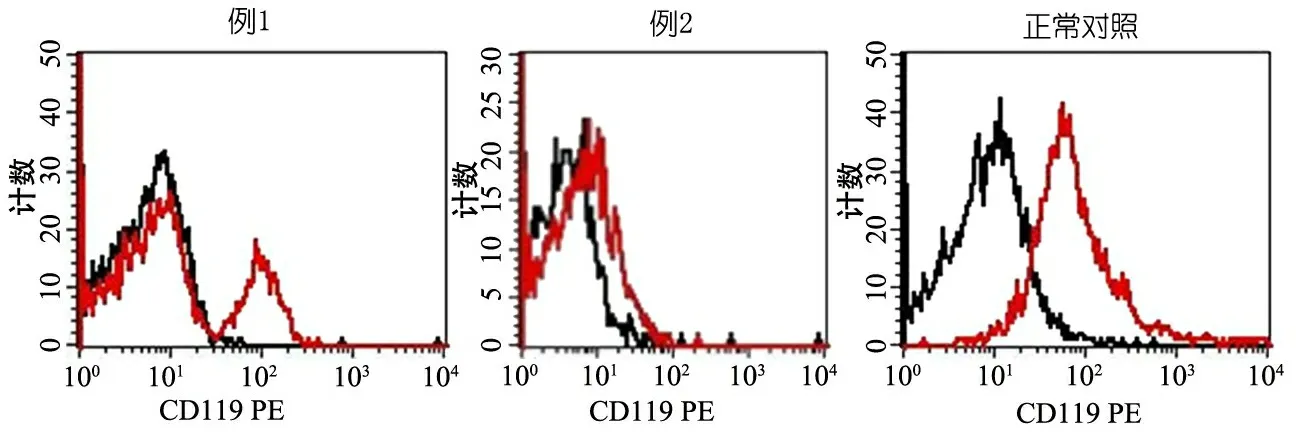
图3 2例患儿及正常人的IFNGR1蛋白表达(流式细胞术)
注 例1 IFNGR1蛋白呈部分表达,例2 IFNGR1蛋白不表达
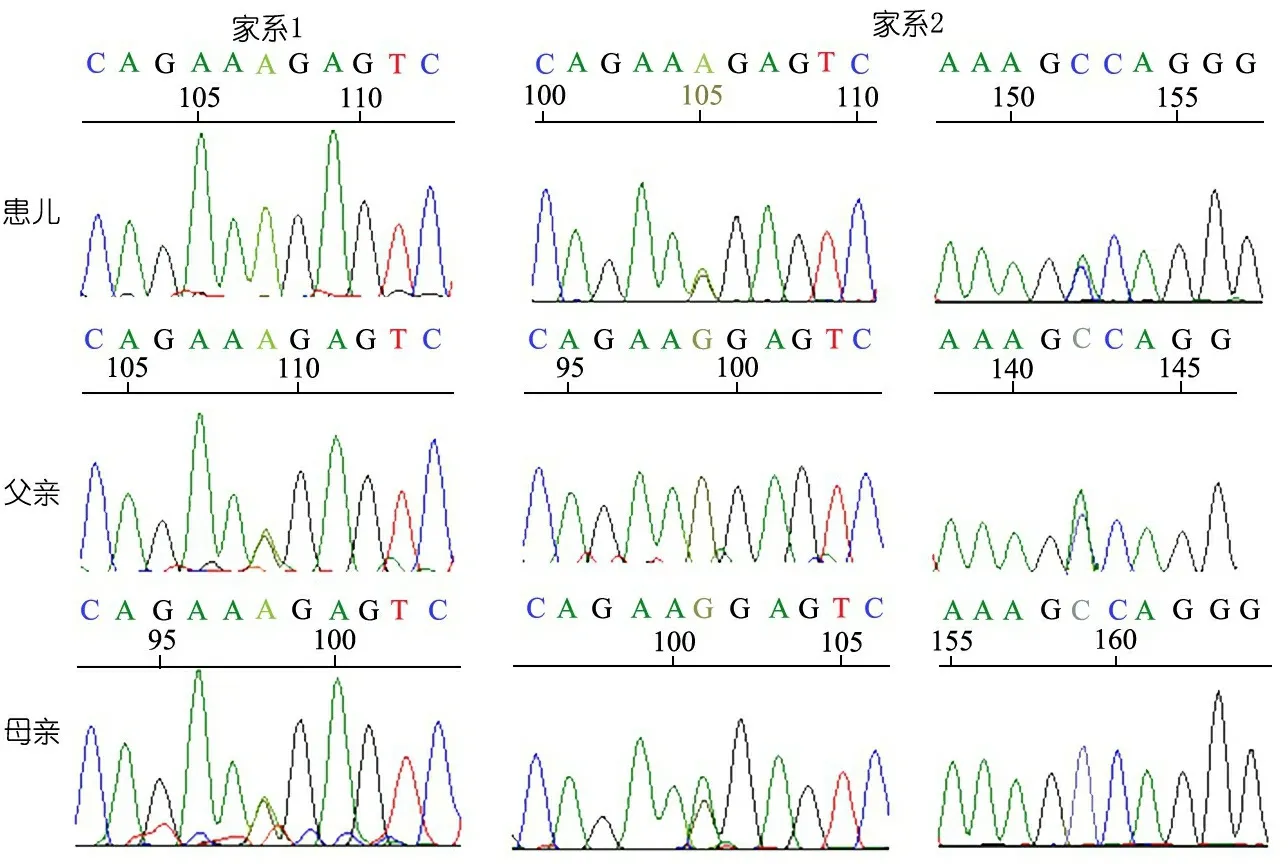
图4IFNGR1基因测序(Sanger法)
注 例1存在c.665 G>A(p.G219R)纯合突变,其父母均为c.665 G>A(p.G219R)杂合突变。例2 存在c.665 G>A(p.G219R)及c.310 C>A(p.A104N)复合杂合突变,其母亲为c.665 G>A(p.G219R)杂合突变,其父亲为c.310 C>A(p.A104N)杂合突变
2 讨论
据WHO统计,2014年全球约9.6亿人口感染了结核杆菌,其中约1 500万人因结核病死亡,预计到2020年每年将有3 600万人死于结核感染[3]。我国结核病发生率居世界第二。接种卡介苗是预防重症结核感染的重要手段之一。卡介苗是一类减毒的活性牛结核分枝杆菌疫苗,将其接种到机体后引起轻度感染,诱导机体产生记忆性T细胞,从而达到预防婴幼儿结核性脑膜炎和播散性结核感染的目的。绝大多数婴儿接种卡介苗后无不良反应,少部分婴儿会发生卡介苗感染,严重者发生播散性感染甚至死亡。我国疾病预防控制中心的调查报告显示,卡介苗严重异常反应率为22.33/100万[4],实际卡介苗感染的发生率可能高于此数据。
宿主自身的免疫缺陷是导致卡介苗病的重要原因。MSMD中,由IFNGR1基因缺陷导致的约占29%[2]。其主要机制为,IFNGR1基因缺陷导致吞噬细胞表面的IFNGR1蛋白表达异常,不能识别T细胞及NK细胞分泌的自然配体IFN-γ,致使吞噬细胞无法被激活而发挥其吞噬分枝杆菌的作用[5]。目前尚无IFNGR1缺陷发病率的相关流行病学数据,国际上报道122例[6],国内仅1例疑似该基因突变的报告[7]。IFNGR1缺陷有常染色体隐性和常染色体显性两种遗传方式。根据IFNGR1蛋白表达水平和IFN-γ产生能力,分为完全型和部分型IFNGR1缺陷(完全型:IFNGR1蛋白不表达,无IFN-γ产生能力;部分型:IFNGR1蛋白部分表达,有一定的IFN-γ产生能力)[8]。完全型多为常染色体隐性遗传,该型IFNGR1缺陷临床表现较为严重,一般在生后早期即发生危及生命的感染,多由低毒力的分枝杆菌引起,IFN-γ治疗效果欠佳,病死率高。至今发现39例该类型患儿[2],IFNGR1突变位点多位于胞外区域,IFNGR1蛋白完全不表达[9, 10]。部分型多为常染色体显性遗传,亦有常染色体隐性遗传,该型IFNGR1缺陷临床表现相对较轻,主要影响胞内信号及受体循环,IFNGR1蛋白可有部分表达及功能[8]。
本文2例女患儿起病年龄较早,均于生后3月龄内出现卡介苗感染的临床表现,早于国外报道(3岁以内起病)[10]。可能与我国作为结核病高发国家,新生儿出生后常规接种卡介苗有关。2例均发生了播散性低毒力分枝杆菌感染,感染部位累及淋巴结、肺部、肠道、中枢和骨髓。文献[2]报道,近1/3的完全型IFNGR1缺陷患儿还易发生其他病原感染,如EB病毒、沙门氏菌、铜绿假单胞菌和李斯特菌等。本文2例患儿均未检测到其他病原感染。
2例患儿的IFN-γ产生能力均低下。例1部分表达IFNGR1蛋白,例2不表达。基因测序发现,2例均为常染色体隐形遗传,例1IFNGR1外显子5上有G219R纯合突变,其父母均为杂合突变,突变位于胞外区域;例2在IFNGR1外显子3和5 上分别存在A104N和G219R错义突变,分别来自其父母,为复合杂合突变。其中例1突变位点已报道[11],例2为新发突变,结合功能实验和蛋白表达,证实其为致病突变。
常染色体隐性遗传完全型IFNGR1突变患儿,由于临床表型较重,常规抗结核治疗效果并不理想,而因基因突变导致与IFN-γ结合能力缺陷,应用IFN-γ治疗也不能改善其症状。有文献[12, 13]报道,通过加用IFN-a激活其他IFN途径替代治疗对此类患儿有效,但亦有文献[14]报道该干预可能有害。有研究对11例常染色体隐性遗传完全型IFNGR1突变患儿进行干细胞移植,移植后1年内死亡5例(4例排异,1例严重感染),移植成功5例,且随访1~6年无明显再发感染[5, 15, 16]。本文例2起病年龄小,播散性感染累及多脏器,IFNGR1蛋白不表达,IFN-γ产生能力低下,结合基因分析,考虑为完全型,予IFN-γ治疗中,目前感染控制尚可,但随访时间较短,疗效需进一步观察。
常染色体隐性遗传部分型IFNGR1缺陷临床表现相对较轻。目前报道的15例此类患儿中,9例发生分枝杆菌感染所致骨髓炎,其他病原还包括肺炎支原体、沙门氏菌和流感嗜血杆菌等,1例发生播散性分枝杆菌感染而死亡[2]。本文病例1发生播散性卡介苗感染,IFNGR1蛋白部分表达,IFN-γ产生能力低下,结合基因测序分析考虑为部分型,予IFN-γ治疗,随访30个月,卡介苗感染控制,停抗痨药未再复发。
综上所述,本文2例IFNGR1突变致MSMD患儿起病年龄早,但均在起病后数年才得到明确诊断,表明国内对该病的认识不足。我院前期的研究[17]表明,大部分卡介苗感染患儿存在免疫缺陷可能。因此,对卡介苗感染的患儿,应进行免疫功能评价,以早期发现免疫缺陷。当常规免疫评估无缺陷时,需考虑是否存在IL12/IFN-γ通路缺陷,结合功能实验、基因检测,有助于诊断该病。
[1] Jouanguy E, Altare F, Lamhamedi S, et al. Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette-Guérin infection. N Engl J Med, 1996, 335(26):1956-1961 .
[2] Bustamante J, Boisson-Dupuis S, Abel L, et al. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin Immunol, 2014, 26(6): 454-470 .
[3] WHO.WHO Tuberculosis Fact Sheet 2015.http://www.who.int/mediacentre/factsheets/fs104/en/. World Heathe Organization 15 A.D. March 20th .
[4] 叶莹, 史鲁斌, 李军, 等. 河南省2009至2014年疑似预防接种异常反应监测分析.郑州大学学报(医学版), 2016,51(2):258-262 .
[5] Rosenzweig SD, Holland SM. Defects in the interferon-gamma and interleukin-12 pathways. Immunol Rev, 2005, 203:38-47 .
[6] de Vor IC, van der Meulen PM, Bekker V, et al. Deletion of the entire interferon-γ receptor 1 gene causing complete deficiency in three related patients. J Clin Immunol, 2016, 36(3): 195-203 .
[7] 胥焕, 蒋利萍, 邢超, 等. 干扰素γ受体1缺陷患儿的临床特征及功能、基因分析,第三军医大学学报,2014, 36(18): 1920-1924 .
[8] Wu UI, Holland SM. Host susceptibility to non-tuberculous mycobacterial infections. Lancet Infect Dis, 2015, 15(8): 968-980 .
[9] Olbrich P, Martínez-Saavedra MT, Perez-Hurtado JM, et al. Diagnostic and therapeutic challenges in a child with complete interferon-γ receptor 1 deficiency. Pediatr Blood Cancer, 2015, 62(11): 2036-2039 .
[10] Dorman SE, Picard C, Lammas D, et al. Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies. Lancet, 2004, 364(9451): 2113-2121 .
[11] Tesi B, Sieni E, Neves C, et al. Hemophagocytic lymphohistiocytosis in 2 patients with underlying IFN-receptor deficiency. J Allergy Clin Immunol, 2015,135:1638-1641 .
[12] Ward CM, Jyonouchi H, Kotenko SV, et al. Adjunctive treatment of disseminated Mycobacterium avium complex infection with interferon alpha-2b in a patient with complete interferon-gamma receptor R1 deficiency. Eur J Pediatr, 2007, 166(9): 981-985 .
[13] Bax HI, Freeman AF, Ding L, et al. Interferon alpha treatment of patients with impaired interferon gamma signaling. J Clin Immunol, 2013, 33(5):991-1001 .
[14] van de Wetering D, van Wengen A, Savage ND, et al. IFN-alpha cannot substitute lack of IFN-gamma responsiveness in cells of an IFN-gammaR1 deficient patient. Clin Immunol, 2011, 138(3): 282-290 .
[15] Bax HI, Freeman AF, Anderson AL, et al. B-cell lymphoma in a patient with complete interferon gamma receptor 1 deficiency. J Clin Immunol, 2013, 33(6): 1062-1066 .
[16] Taramasso L, Boisson-Dupuis S, Garre ML, et al. Pineal germinoma in a child with interferon-gamma receptor 1 deficiency. Case report and literature review. J Clin Immunol, 2014, 34(8): 922-927 .
[17] Ying W, Sun J, Liu D, et al. Clinical characteristics and immunogenetics of BCGosis/BCGitis in Chinese children: a 6 year follow-up study. PLoS One, 2014, 9(4): e94485
Two Mendelian susceptibility to mycobacterial diseases patients with novel IFNGR1 gene mutations
YINGWen-jing,LIUDan-ru,SUNJin-qiao,WANGWen-jie,YUYe-heng,WANGXiao-chuan
(DepartmentofClinicalImmunology,Children'sHospitalofFudanUniversity,Shanghai201102,China)
WANG Xiao-chuan, E-mail: xchwang@shmu.edu.cn
ObjectiveIn this study, two Mendelian susceptibility to mycobacterial diseases (MSMD) patients with novelIFNGR1 mutation were reported. MethodsClinical manifestations of two patients withIFNGR1 mutation were reviewed. The IFN-γ level was analyzed by ELISA, the IFNGR1protein expression was detected by flow cytometry. Mutation analysis inIFNGR1 was performed by Sanger sequencing.Results①Clinical manifestations: In 2 patients, BCG disease was found during 3 months after birth, and the draining lymphadenectasis after BCG vaccination was the onset feature. The BCG infection was gradually disseminated to lung, intestinal tract, central nervous system and bone. The diagnosing age of the 2 patients was 4 and 6 years old, respectively. Routine immune function evaluation (lymphocyte subgroup, immunoglobulin, DHR analysis, complement) was normal. ②The two patients showed significantly lower IFN-γ concentrations in the supernatant after stimulation with medium alone, and IFN-γ concentrations did not significantly increase after stimulation with LPS or with LPS plus IL-12. ③IFNGR1 protein expression was very low in the two patients. ④Gene sequencing: Case 1 had a c.665G>A (p.G219R) homozygous mutation, whose parents were both with c.665G>A (p.G219R) heterozygotes mutation. Case 2 had c.665G>A (p.G219R) and c.310 C>A (p.A104N) compound heterozygous mutation, and her mother had c.665G>A (p.G219R) heterozygous mutation and her father had c.310 C>A (p.A104N) heterozygous mutation. The mutation A104N was novel. ⑤Treatment: Both patients were suffered recurrent mycobacteria infection before diagnosis. While treated with IFN-γ after diagnosis, the BCG infection was controlled without any adverse reactions.ConclusionIFNGR1 gene mutation can cause MSMD. When the routine immunological evaluation of patients with BCG disease is normal, it is necessary to consider the possibility of MSMD. The detection of protein expression and gene analysis are helpful to the diagnosis of the disease.
Mendelian susceptibility to mycobacterial diseases;IFNGR1 gene; BCG disease
2017-08-01
2017-08-15)
(本文编辑:张崇凡,孙晋枫)
上海市科学技术委员会西医引导项目:14411965400
复旦大学附属儿科医院临床免疫科 上海, 201102
王晓川,E-mail: xchwang@shmu.edu.cn
10.3969/j.issn.1673-5501.2017.04.011
