高效液相色谱法测定复方锌布颗粒剂中马来酸氯苯那敏的含量及含量均匀度
2017-09-11冯璐
冯璐
(江苏省泰州市食品药品检验所,江苏 泰州 225300)
·检验检测·
高效液相色谱法测定复方锌布颗粒剂中马来酸氯苯那敏的含量及含量均匀度
冯璐
(江苏省泰州市食品药品检验所,江苏 泰州 225300)
目的 建立测定复方锌布颗粒剂中马来酸氯苯那敏含量及含量均匀度的高效液相色谱(HPLC)法。方法 色谱柱采用 Capcell PAK C18柱(250 mm×4.6 mm,5 m),流动相为0.05 mol/L KH2PO4(含0.1%三乙胺,以磷酸调节 pH至2.5)-乙腈(80∶20),流速为1.0 mL/min,柱温为35℃,检测波长为263 nm。结果 马来酸氯苯那敏质量浓度在0.006 356~0.203 4 g/L(r=1.000 0)范围内与峰面积线性关系良好,平均回收率为100.17%,RSD为0.97%(n=9)。结论 该方法简便、快捷,可有效、准确地测定马来酸氯苯那敏的含量及含量均匀度,可用于复方锌布颗粒剂的质量控制。
高效液相色谱法;马来酸氯苯那敏;含量均匀度
复方锌布颗粒剂为儿童用感冒药,用于缓解普通感冒或流行性感冒引发的发热、头痛、四肢酸痛、鼻塞、流涕、打喷嚏等症状[1-2],主要成分包括葡萄糖酸锌、布洛芬及马来酸氯苯那敏。目前,《中国药典》尚未收载该品种,现行标准采用紫外-可见分光光度法测定马来酸氯苯那敏的含量[3]。2015年版《中国药典(二部)》中收录的马来酸氯苯那敏采用高氯酸滴定法测定含量。现行标准中,测定方法涉及使用有毒试剂苯,其具有挥发性、高毒性与致癌性;药典中,涉及使用腐蚀性强的高氯酸。试验流程均复杂,结果偏差大。此外,复方锌布颗粒剂中马来酸氯苯那敏规格仅为2 mg,用量极少,不易分散均匀,过量使用可能出现泌尿系统不良反应和精神症状[4]。马来酸氯苯那敏存在一定的紫外吸收,参考文献[5-11],本研究中通过优化色谱条件,采用高效液相色谱法测定马来酸氯苯那敏的含量及含量均匀度,方法简便、快速、灵敏度高、误差小、重复性好、专属性强、结果准确,可用于该产品的质量控制。现报道如下。
1 仪器与试药
1.1 仪器
Waters e2695/2996型高效液相色谱仪(日本岛津公司);S20型精密pH计(梅特勒-托利多仪器上海有限公司);XS105DU型电子天平(梅特勒-托利多公司)。
1.2 试药
马来酸氯苯那敏对照品(中国食品药品检定研究院,批号为 100047-201507);乙腈为色谱纯(Scharlab S.L.);水为自制超纯水;其余试剂均为分析纯;复方锌布颗粒剂均为市售品,系7个厂家共8批次样品(批号分别为20160827,20160527,20160219,20160905,20161103,20150106,20150212,20150309),其中葡萄糖酸锌、布洛芬、马来酸氯苯那敏的规格分别为100 mg,150 mg,2 mg。
2 方法与结果
2.1 色谱条件
色谱柱:Capcell PAK C18柱(250 mm×4.6 mm,5 μm);流动相:0.05 mol/L KH2PO4(含0.1%三乙胺,以磷酸调节pH至2.5)-乙腈(80∶20);流速:1.0 mL/min;柱温:35℃;检测波长:263 nm;进样量:20 μL。
2.2 溶液制备
对照品溶液:取干燥至恒重的马来酸氯苯那敏对照品适量,精密称定,置容量瓶中,加流动相适量,使马来酸氯苯那敏溶解,再用流动相稀释至刻度,得质量浓度为1.0 g/L的马来酸氯苯那敏对照品贮备液。精密量取1 mL马来酸氯苯那敏对照品贮备液,置25 mL容量瓶中,用流动相稀释至刻度,摇匀,即得。
供试品溶液:精密称取供试品适量(约相当于马来酸氯苯那敏2 mg),置50 mL容量瓶中,加入适量流动相溶解并稀释至刻度,混合,摇匀,过滤,取续滤液,即得。
阴性对照品溶液:按复方锌布颗粒剂的处方,制备缺马来酸氯苯那敏的样品,按供试品溶液的制备方法制备成阴性对照溶液。
2.3 方法学考察
系统适用性试验:按拟订色谱条件,分别精密量取对照品溶液、供试品溶液以及阴性对照品溶液各20 μL,注入液相色谱仪,记录图谱,见图1。马来酸氯苯那敏的保留时间为9.1min,理论板数按马来酸氯苯那敏峰计为10 000。

图1 高效液相色谱图
检测波长选择:取马来酸氯苯那敏对照品溶液,于200~400 nm波长范围内扫描,结果在263 nm波长处有最大吸收,详见图2。故选择263 nm作为测定波长。

图2 马来酸氯苯那敏紫外扫描图
线性关系考察:精密吸取2.2项下对照品贮备液,制备质量浓度为0.006 25,0.0125,0.025,0.05,0.1,0.2 g/L的系列溶液,按拟订色谱条件测定马来酸氯苯那敏峰面积,以马来酸氯苯那敏质量浓度(X)为横坐标、峰面积(Y)为纵坐标绘制标准曲线,得回归方程 Y=2.24×107X-9 401.70,r=1.000 0(n=6)。结果表明,马来酸氯苯那敏质量浓度在0.006 356~0.203 4 g/L范围内与峰面积线性关系良好。详见图3。

图3 马来酸氯苯那敏标准曲线
精密度试验:取2.2项下对照品溶液20 μL,连续进样6次,测得马来酸氯苯那敏峰面积的 RSD为0.6%(n=6),表明仪器精密度良好。
重复性试验:取同一批供试品(批号为20160219),按2.2项下方法平行制备6份供试品溶液,依法进样测定。结果测得马来酸氯苯那敏的含量为101.2%,RSD为1.5%(n=6),表明方法重复性良好。
稳定性试验:取同一供试品溶液,分别于0,2,4,8,16,24 h时进样测定。结果峰面积值基本无变化,RSD为0.6%(n=6),表明供试品溶液在24 h内稳定。
检测限与定量限测定:取20 μL马来酸氯苯那敏对照品溶液注入液相色谱仪,计算信噪比(S/N)。结果马来酸氯苯那敏的检测限为2.59×10-4g/L(S/N=3),定量限为9.38×10-4g/L(S/N=10)。

表1 马来酸氯苯那敏加样回收试验结果(n=9)
加样回收试验:精密称取已知含量的同一批供试品(批号为20160219)9份,置容量瓶中,分别加入对照品溶液适量,加入流动相定容,过滤,取续滤液。按拟订色谱条件进样测定。结果见表1。
2.4 样品含量及含量均匀度测定
按2.2项下方法制备供试品溶液,依次对8批次不同厂家不同批号的样品进行含量均匀度测定。由于药品生产批号不同,相关公式分别参考2010年版《中国药典》规定。结果见表2。
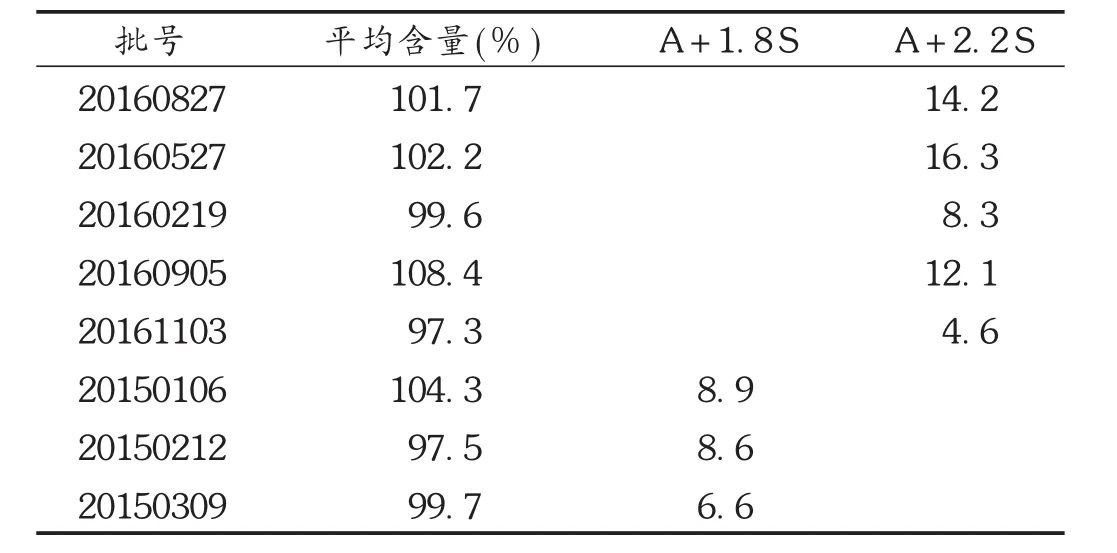
表2 8批样品含量及含量均匀度测定结果
3 讨论
3.1 流动相选择
尝试采用不同比例0.05 mol/L KH2PO4-乙腈,最终采用了0.05 mol/L KH2PO4(加入0.1%的三乙胺,再用磷酸调pH至2.5)-乙腈(80∶20)作为流动相,主峰出峰时间约为9 min,比较合适。曾适当增加乙腈,结果发现出峰时间过快,且在使用流动相作为溶剂制备供试品溶液时,可导致乙腈用量偏大,故最终确定比例为80∶20。三乙胺作为一种缓冲盐和改性剂,可减少样品中组分的峰拖尾现象,也可改善主峰拖尾的现象。
3.2 含量及含量均匀度测定结果
复方锌布颗粒剂中对于马来酸氯苯那敏含量的标准为标示量的90.0%~110.0%,结果表明,此8批样品测得的平均含量均符合规定。对于标准中并未规定的含量均匀度,有2批次样品的结果都在边缘,有可能企业在生产投料过程中的准确度及混料等过程中有一定问题,建议质量标准中增加对马来酸氯苯那敏含量均匀度的检查,可在一定程度上反映产品的质量。
[1]兰 文,李昌亮.高效液相色谱法测定复方锌布颗粒中布洛芬的溶出度[J].中南药学,2015,13(12):1314-1317.
[2]吴 琳.复方锌布颗粒剂扑尔敏含量测定方法的比较[J].中华中西医学杂志,2008,6(8):75-76.
[3]WS1-(X-092)-95Z,中华人民共和国卫生部药品标准[S].
[4]郝晶晶,李 伟,李海亮,等.小儿氨酚黄那敏颗粒中对乙酰氨基酚及马来酸氯苯那敏含量及含量均匀度考察[J].中国药师,2013,16(1):38-39.
[5]谢 华,傅 萍,张悦杨,等.HPLC测定复方氨酚烷胺片中的对乙酰氨基酚、咖啡因和马来酸氯苯那敏[J].华西药学杂志,2012,27(3):307-308.
[6]黄诺嘉,罗丽娟.HPLC测定扑感片中对乙酰氨基酚和马来酸氯苯那敏的含量[J].中成药,2007,29(10):1462-1465.
[7]阚微娜,杨宏伟.HPLC测定复方氨肽素片中氨茶碱与马来酸氯苯那敏的含量及含量均匀度[J].中国现代应用药学,2015,32(7):835-838.
[8]王 烽,朱祥松.HPLC法测定复方锌布颗粒剂中布洛芬和马来酸氯苯那敏的含量[J].中国药品标准,2008,9(2):156-158.
[9]董 丽,孙祥德,李 琴.HPLC法同时测定复方锌布颗粒剂中3种组分的含量[J].药物分析杂志,2010,30(10):1883-1886.
[10]陈晓亮,汪 旭.HPLC法同时测定克敏乳膏中3种成分的含量[J].中国药房,2016,27(33):4725-4727.
[11]黄玉昌,曾庆伟,吴芳花,等.高效液相色谱法测定复方锌布颗粒中马来酸氯苯那敏含量[J].中国药业,2014,23(1):31-33.
Determination of Content and Content Uniformity of Chlorphenamine Maleate in Compound Zinc Gluconate and Ibuprofen Granules by HPLC
Feng Lu
(Taizhou Institute For Food and Drug Control,Taizhou,Jiangsu,China 225300)
Objective To establish an HPLC method for the determination of content and content uniformity of chlorphenamine maleate in Compound Zinc Gluconate and Ibuprofen Granules.Methods The Capcell PAK C18column(250 mm×4.6 mm,5 μm)was adopted,the mobile phase was 0.05 mol/L KH2PO4(containing 0.1% triethylamine three,pH was adjusted to 2.5 by phosphoric acid)-acetonitrile(80∶20),the flow rate was 1.0 mL/min,the column temperature was 35℃,the wavelength was 263 nm.Results There was a good linear relationship within the range of 0.006 356-0.203 4 g/L(r=1.000 0).The average recovery was 100.17%,RSD was 0.97%(n=9).Conclusion The method is convenient,rapid,effective and accurate for the determination of content and content uniformity of chlorphenamine maleate,which can be used in the quality control of Compound Zinc Gluconate and Ibuprofen Granules.
HPLC;chlorphenamine maleate;content uniformity
2017-04-10)
10.3969/j.issn.1006-4931.2017.15.006
冯璐,女,硕士研究生,研究方向为药物分析,(电子信箱)wuyuyd@163.com。
R927.2
A
1006-4931(2017)15-0020-03
