Dissociative Photoionization of 1,4-Dioxane with Tunable VUV Synchrotron Radiation
2017-09-03MingWangJunChenWeifeiFeiZhaohuiLiYepengYuXuanLinXiaobinShanFuyiLiuLiusiSheng
Ming Wang,Jun Chen,Wei-fei Fei,Zhao-hui Li,Ye-peng Yu,Xuan Lin,Xiao-bin Shan, Fu-yi Liu,Liu-si Sheng
National Synchrotron Radiation Laboratory,University of Science and Technology of China,Hefei 230029,China
(Dated:Received on March 11,2017;Accepted on April 11,2017)
Dissociative Photoionization of 1,4-Dioxane with Tunable VUV Synchrotron Radiation
Ming Wang,Jun Chen,Wei-fei Fei,Zhao-hui Li,Ye-peng Yu,Xuan Lin,Xiao-bin Shan, Fu-yi Liu∗,Liu-si Sheng
National Synchrotron Radiation Laboratory,University of Science and Technology of China,Hefei 230029,China
(Dated:Received on March 11,2017;Accepted on April 11,2017)
The photoionization and photodissociation of 1,4-dioxane have been investigated with a reflectron time-of- flight photoionization mass spectrometry and a tunable vacuum ultraviolet synchrotron radiation in the energy region of 8.0−15.5 eV.Parent ion and fragment ions at m/z 88,87,58,57,45,44,43,41,31,30,29,28 and 15 are detected under supersonic conditions.The ionization energy of DX as well as the appearance energies of its fragment ions C4H7O2+,C3H6O+,C3H5O+,C2H5O+,C2H4O+,C2H3O+,C3H5+,CH3O+, C2H6+,C2H5+/CHO+,C2H4+and CH3+was determined from their photoionization efficiency curves.The optimized structures for the neutrals,cations,transition states and intermediates related to photodissociation of DX are characterized at the B3LYP/6-31+G(d,p) level and their energies are obtained by G3B3 method.Possible dissociative channels of the DX are proposed based on comparison of experimental AE values and theoretical predicted ones.Intramolecular hydrogen migrations are found to be the dominant processes in most of the fragmentation pathways of 1,4-dioxane.
1,4-Dioxane,VUV photoionization mass spectra,Appearance energy,Dissociation channel,G3B3
I.INTRODUCTION
1,4-Dioxane(DX)can be easily found in nature as an intermediate in many chemical pathways[1−3].It has a wide range of applications in paints[1,2],textile and dye industries[2].On the other hand,DX is also well known to induce kidney failure,liver damage [4,5]and even human carcinogen.Recently,DX,a signi ficant representative of cyclic ethers,has also been con firmed hard to degrade[6,7],which will contribute to the concentration of DX in the atmosphere[8,9].DX can undergo further transformation or degradation with OH radicals and halogen atoms(such as Br,Cl)[9], then transform into low-volatility oxygenated organic compounds,which may contribute to secondary organic aerosol formation under atmospheric conditions.Hence, a better understanding of the unimolecular chemistry of DX is clearly desirable.
Up to now,a lot of methods[10−19]have been devoted to the unimolecular chemistry of DX.As an important physical property,the ionization energy(IE)ofDX wasdeterminedtobeinthe rangeof9.058−9.39 eV by photoionization (PI) spectroscopy[19],photoelectron-photoion-coincidence (PEPICO)spectroscopy[12],mass analyzed threshold ionization(MATI)spectrum[18]and photoionization mass spectrometry(PIMS)[15].The appearance energy(AE)values of fragment ions investigated by different techniques also show considerable discrepancies. These large deviations may be enough to remind researchers to re-investigate the details of fragmentation patterns.In the case of dissociating channels,Fraser-Monteiro and co-workers[12]discussed the possible structures and the heats of formation for fragment ions from DX by using PEPICO.Then,they concluded that ionic DX trend to produce fragment ions and neutrals by simple bond cleavages.Zou et al.[15]discussed the possible fragmentation channels and structures of some fragment ions and argued that the rearrangement processes were involved in the dissociation of DX,not just involved breaking bonds.It should be noted that AE values derived from the previous PIMS suffered from poor signal to noise ratio.Apart from these experimental studies,there are a few theoretical reports describing the dissociative photoionization process of DX.Lam et al.[20]studied the detailed mechanism of C3H6O+isomers(m/z=58)fragmented from DX cation and Hudson et al.[21]explored the formation behaviors of C2H6+(m/z=30)and C2H5+(m/z=29)from CH2CH2OCH2+.
As mentioned above,despite considerable experime-ntal studies performed on cationic DX,the IE,AE values as well as some formation pathways of the fragment ions,still have not been de finitely con firmed.Thus, more evidences based on real-time analysis and isomeric selectivity are needed to fully understand the dissociative mechanism of DX.This may be obtained by using tunable VUV-PIMS technique[22−27].This approach features several advantages.First,the molecular-beam reduces collision effects and allows unstable intermediates to be isolated.Furthermore,the high-energy resolution and tunability of the synchrotron radiation minimize fragmentation and allows different isomers to be distinguished[26,27].Indeed,the experimental method employed in this work was also successfully employed in the previous studies[22−25].In this work,we report a quantitative study on the photoionization and dissociative photoionization of DX on the basis of VUV-PIMS experiments and theoretical calculations.
II.EXPERIMENTAL AND THEORETICAL METHODS
A.Experimental methods
Thewholeexperimentswereperformedatthe atomic and molecular physics beamline(BL09U)at the National Synchrotron Radiation Laboratory(NSRL) Hefei,China. Thesynchrotron radiationbeam from an undulator of the 800 MeV electron storage ring of the NSRL was monochromatized with a 6-m length monochromator equipped with a grating(370 lines/mm),which covers the photon energy range from 7.5 eV to 22.5 eV.The absolute photon of monochromator is precisely calibrated with the known IEs of inert gases.The energy resolution(E/∆E)of photons at 15.9 eV is measured to be 9 meV(full width at half maximum)[28],with an average photon flux~1012photons/s.The monochromatic VUV radiation, the supersonic molecular beam and the PIMS are mutually perpendicular.The setup consists of seven electrostatic lenses that focus and accelerate the ions from the region of interaction to the home-made TOF-MS with a mass resolution∆M/M of~800.An overview of the experimental setups have been reported in Refs.[29, 30].For each target molecules,the experiments consist of the acquisition of the mass spectra at designated photon energies and the measurement of the PIE curves of the selected ions.All the PIE curves are obtained by integrating over the peaks at each photon energy and normalized by the photon flux recorded by a photodiode.The experimental AEs,corresponding to the onset of the ion signal,can be determined from PIE curves by fitting straight lines to the background and to the ion signal in the threshold region,which has been proven to be very useful[22−25,31−34].
The liquid sample of DX(Alfa-Aesar,99%purity) without further puri fication is carried by Ar(1.5 atm, 99.99%purity)and then expanded into the ionization chamber through a nozzle with a diameter of 70µm. The beam consisting of neutral monomeric DX then passes through a skimmer with diameter of 1 mm to form a continuous supersonic molecular beam before it reaches the photoionization region.The skimmed molecular beam is detected by the VUV radiation in the ionization region of a TOF-MS.In view of the supersonic jet expansion conditions,the thermal energy distribution of parent molecule is not taken into account in data processing.During the experiment,Ar is used as the filter gas with an operating pressure of 6 Torr to effectively suppress higher harmonics of the undulator[32].
B.Theoretical calculation
In the process of photoionization and dissociative photoionization of a molecule M by monochromatic radiation of energy hν,the ionic fragment m0+,and several neutral fragments miare produced as the following equation.
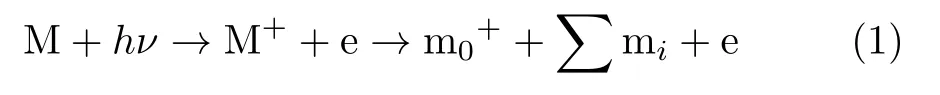
The adiabatic IE of M is determined by using Eq.(2) and the adiabatic AE of m0+is calculated by Eq.(3):
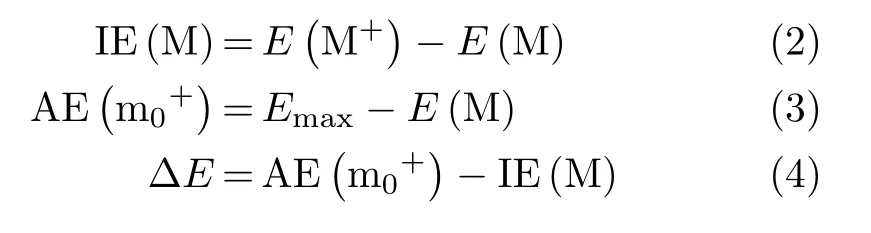
where Emaxrefers to the highest energy barrier involved in the formation pathway of corresponding ionic fragment m0+.E(M+)and E(M)represent the absolute energy of ionic and neutral precursor,respectively.The energies to form related products are calculated by subtracting adiabatic IE of the parent molecule from its adiabatic AE,namely Eq.(4).
To obtain IE for M and AE values for various fragment ions,we performed calculations of single-point energies for reactants,products,intermediates and transition states at G3B3 level and the calculated results are given in Table S1 in supplementary materials.In this theory,prior to energetic calculations,the geometry of a species is optimized at the B3LYP/6-31+G(d,p)level with the Gaussian 09 programs[35].The calculated values of the IE,AE,and∆E for possible formation pathways in the dissociative photoionization of DX are summarized in Table I as well as our experimental values.
III.RESULTS AND DISCUSSION
A.Experimental measurements
The VUV photoionization mass spectra of DX at photon energies of 15.50,11.51,and 9.50 eV areshown in FIG.1.At the photon energy of 15.50 eV, ions at m/z 15,28,29,30,31,41,43,44,45,57, 58,87 and 88 can be recognized as CH3+,C2H4+, C2H5+/CHO+,C2H6+,CH3O+,C3H5+,C2H3O+, C2H4O+,C2H5O+,C3H5O+,C3H6O+,C4H7O2+and molecular ion C4H8O2+,respectively.This is well consistent with the data from the NIST database[36].The signal at m/z=18 is neglected because it is water from the background of setup.With photon energy decreasing to 11.51 eV,a few ion signals C2H4O+,C2H5O+, C3H5O+,C3H6O+,C4H7O2+,and C4H8O2+can be distinguished clearly.When the photon energy is further reduced to 9.50 eV,only parent ion can be detected.
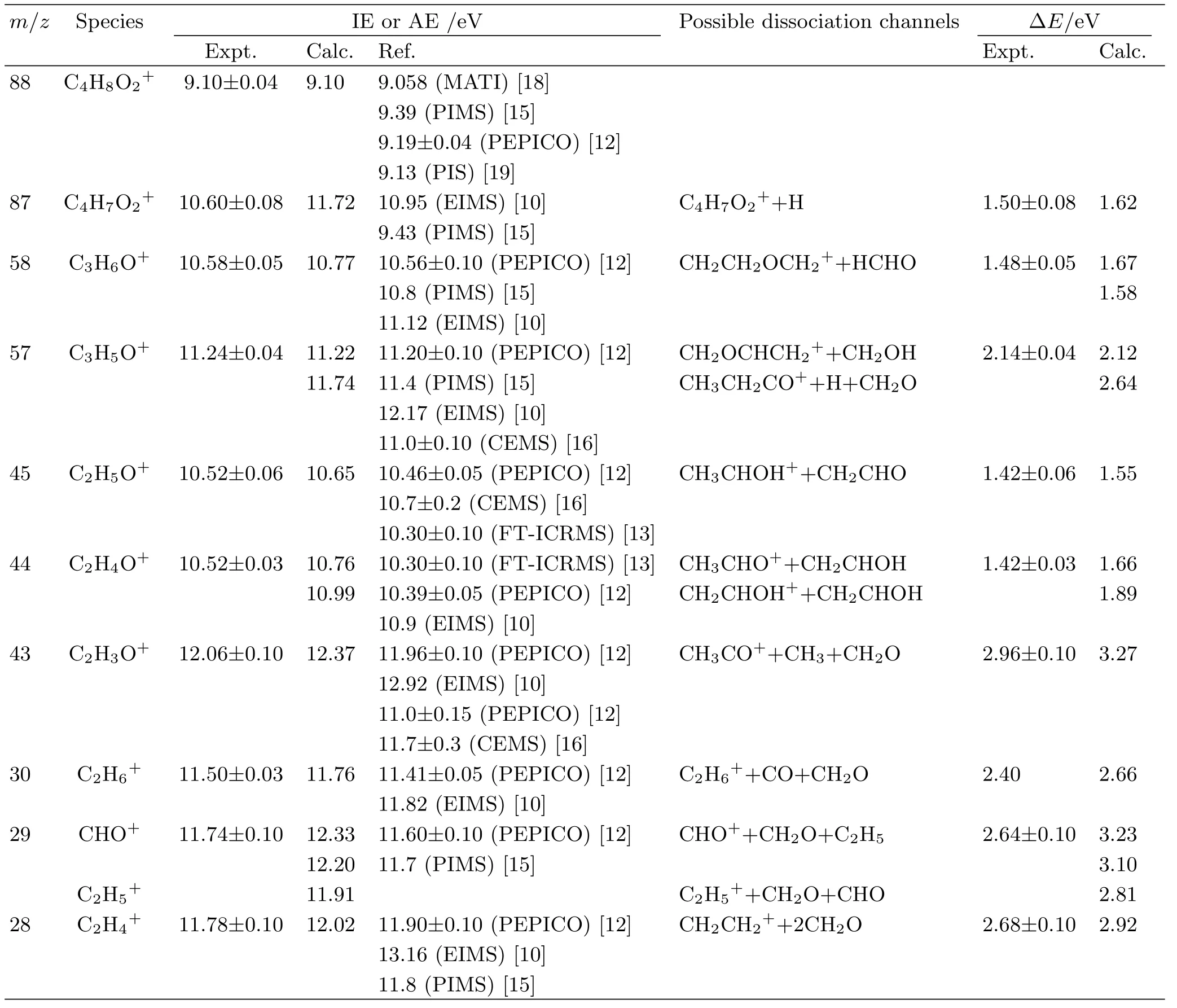
TABLE I Experimental and calculated values of IE and AEs and∆E(energies to form products)for possible dissociative channels in the dissociative photoionization of 1,4-dioxane.
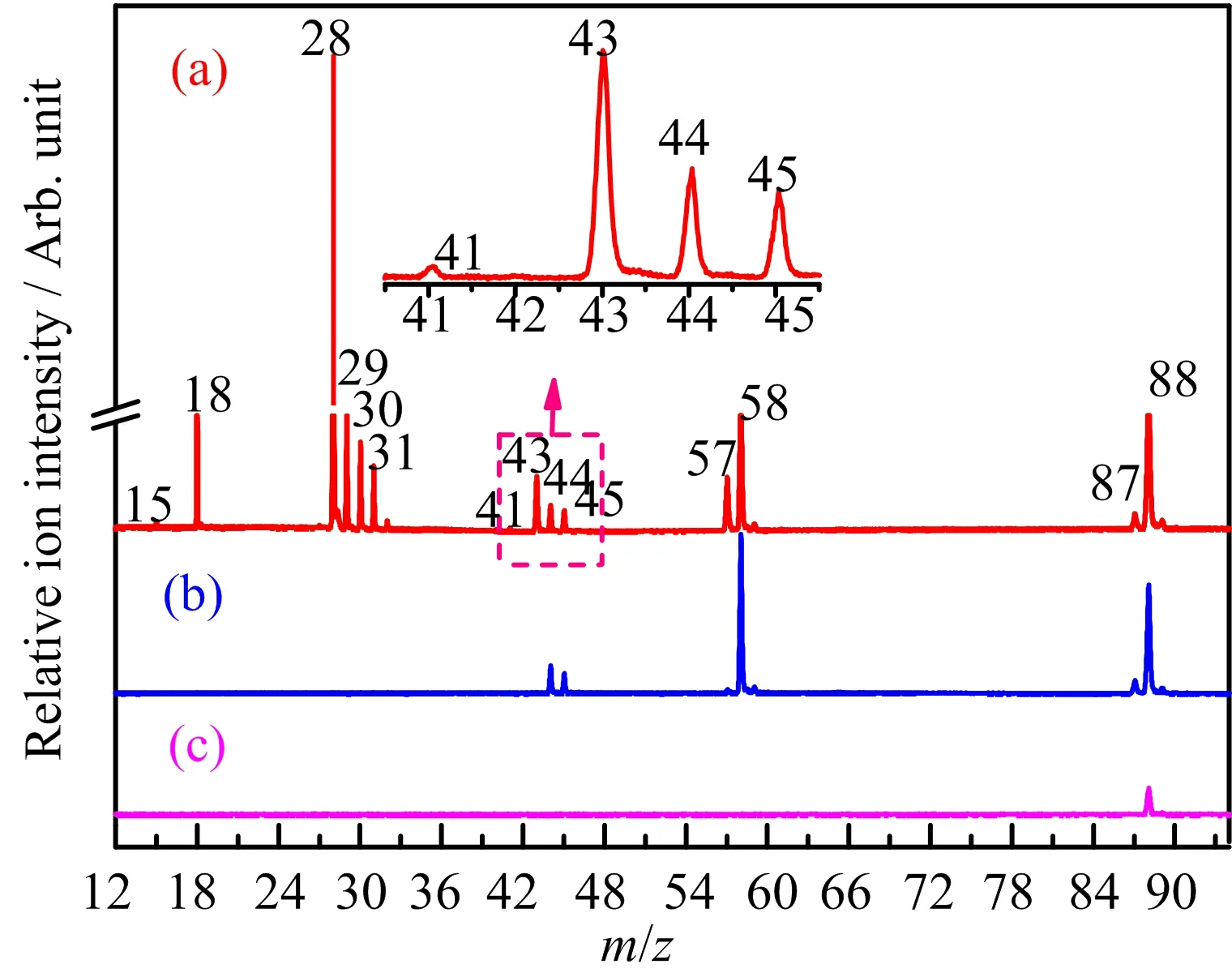
FIG.1 Photoionization mass spectra of 1,4-dioxane at (a)15.50,(b)11.51,and(c)9.50 eV.The insert shows expanded mass spectra between the m/z range from 41 to 45.
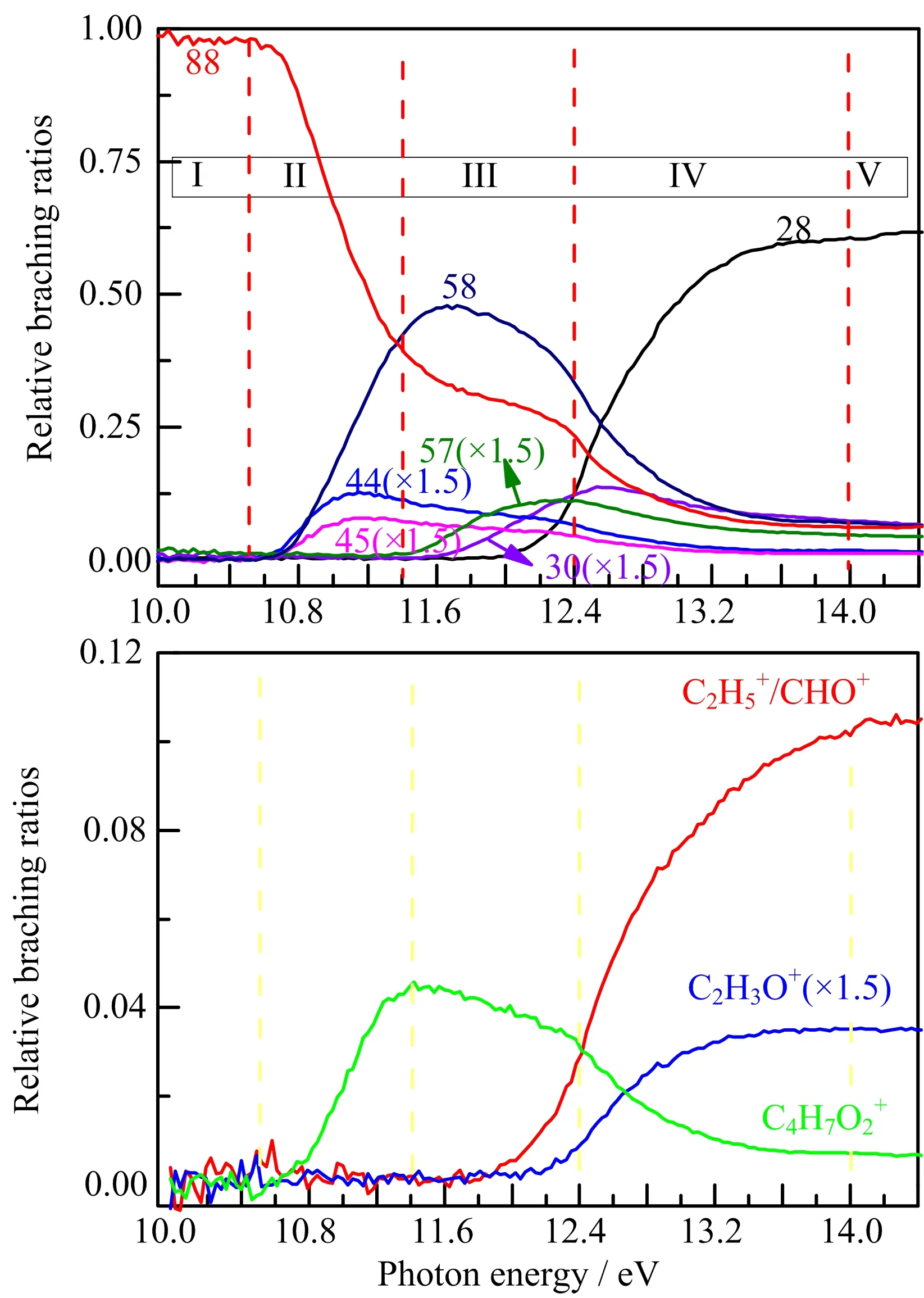
FIG.2 Relative branching ratio of parent ion(C4H8O2+) and observed major fragment ions including C4H7O2+, C3H6O+ (m/z=58), C3H5O+ (m/z=57), C2H5O+(m/z=45),C2H4O+ (m/z=44),C2H3O+ (m/z=30), C2H6+(m/z=28),CHO+/C2H5+,and C2H4+,derived from normalized photoionization efficiency curves.
The relative branching ratio curves of various charged fragment are shown in FIG.2. For the sake of comparison,we arti ficially divide them into five regions.In area(I),the C4H8O2+remains to be the only ion from its onset to~10.5 eV.In area(II), the C4H8O2+decreases abruptly while fragment ions C4H7O2+,C3H6O+,C2H5O+and C2H4O+rise and their onsets cross at~10.5 eV.This clearly indicates that these four fragment ions come from the directly dissociation of the parent ion. Obviously,C3H6O+dominates the ion population until~12.6 eV,corresponding to a major dissociation channels.The similar AE values and shape of branching ratio curves of C2H5O+and C2H4O+may indicate a common intermediate.When it comes to area(III),C4H8O2+decreases slowly along with the signi ficant reduction of C3H6O+. Meanwhile,the ionic species C3H5O+and C2H6+begin to increase,indicating that these species may originate from C4H7O2+or C3H6O+or even both.Analogous analysis can also be used for fragment ions C2H3O+, CHO+/C2H4+and C2H4+in area(IV).In the energy range of 12.4−14.0 eV,C4H8O2+and C3H6O+decrease rapidly,as other fragment ions,such as C2H3O+, CHO+/C2H4+and C2H4+are produced more quickly. Moreover,the dissociation channel to produce C2H4+predominates above 12.6 eV.As for area(V),all the relative branching ratio values seem to remain stable.
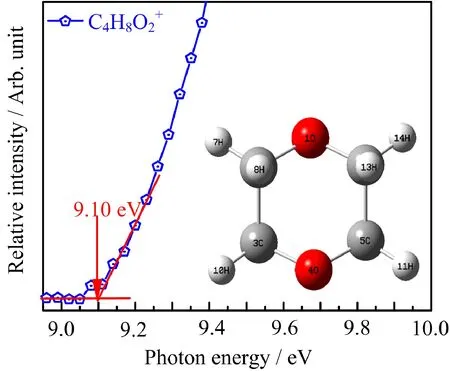
FIG.3 Normalized photoionization efficiency curves of the parent ion C4H8O2+.The ionization energy is determined to be(9.10±0.04)eV.The inset picture is the DFT optimized lowest energy structure for 1,4-dioxane cation.
The PIE curves of the parent ion and its main fragment ions are shown in FIG.3 and FIG.4,respectively.For the parent ion,our measured IE value of 9.10±0.04 eV is consistent with the experimental determination of 9.058 eV obtained from MATI measurement[18]and calculated value of 9.10 eV at G3B3 level.The geometries of parent ion and neutral DX at B3LYP/6-31+G(d,p)level are very similar because of symmetry restrictions that the oxygen lone pairs are primarily p-type orbitals and are essentially nonbonding.This is the reason why the PIE curve of C4H8O2+exhibits a relatively sharp onset to some extent.
B.Dissociation mechanisms
1.Formation pathway of C4H7O2+
DX has a chair conformation with a center of symmetry in the gas phase and possesses the point group of D2h[37].That is to say there are two types of C−H bonds(for example:C6−H14 point to the plane and C6−H13 point out of the plane)in DX.The fragmentation channel attributable to the loss of an H atom in DX may occur via two distinguishable fragmentation pathways.In one case,the process involves the rupture of the C6−H13 bond with an energy barrier of 1.62 eV (TS1-1),resulting in a six-membered ring with double bond structure(P2).The calculated AE of C4H7O2+in this channel(named channel 1a in the following)is 10.72 eV,consistent well with our experimental value of(10.60±0.08)eV.The other case,named channel 1b, involves the cleavage of C6−H14 bond overcoming an energy barrier of 2.53 eV,forming the final products P2 and H atom.These measurements of AE values and energy barriers show that channel 1a is favored.
2.Formation pathway of C3H6O+
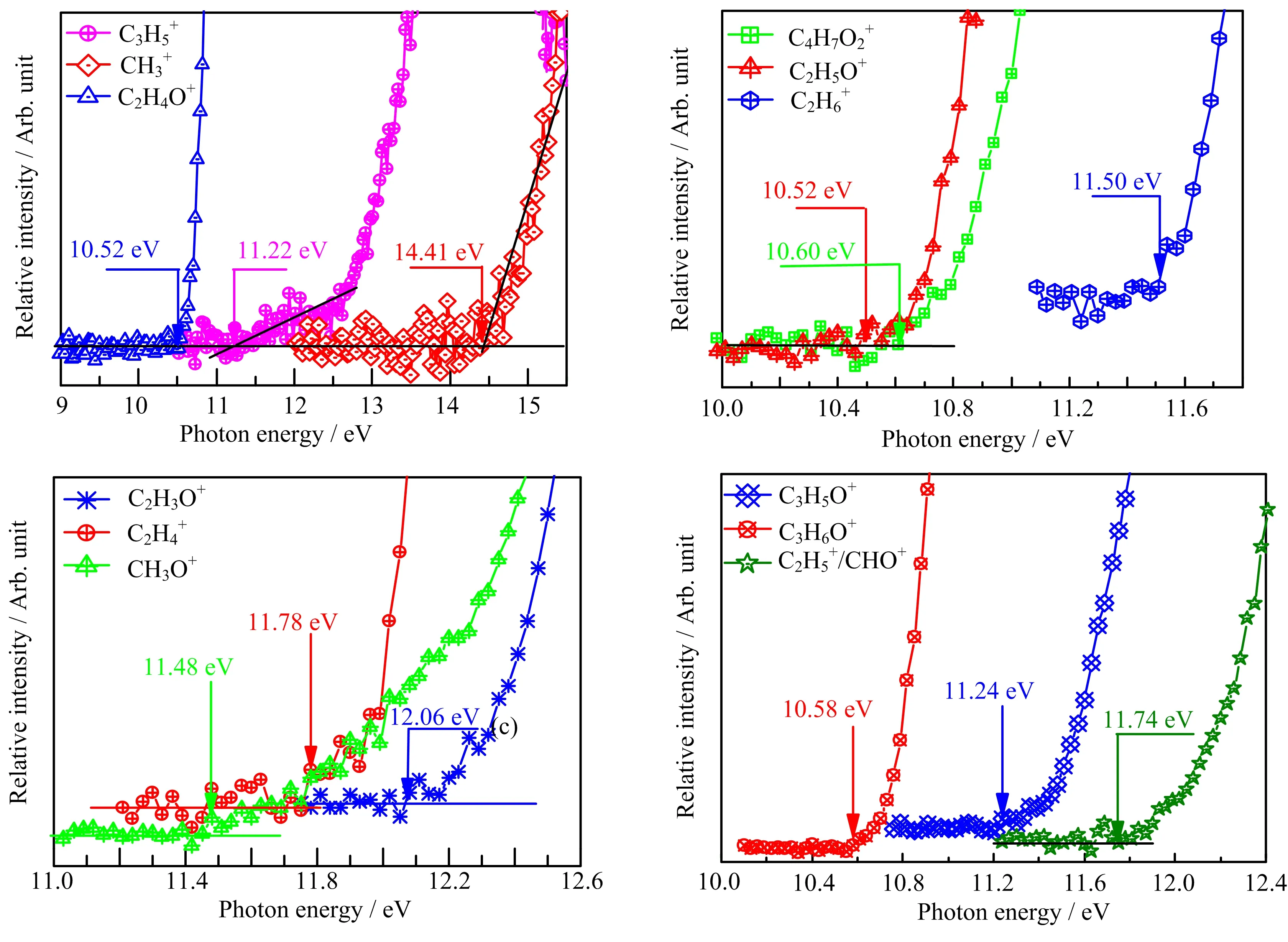
FIG.4 Normalized photoionization efficiency curves and appearance energy values of fragment ions measured in the dissociative photoionization of 1,4-dioxane.
As the dominant fragment product of C4H8O2+between 10.9−12.5 eV,the fragment ion C3H6O+has been con firmed to be a mixture of CH2CH2OCH2+as major component(90%−95%)and CH3OCHCH2+as a minor component(5%−10%)of the C3H6O+ions[14, 20].Lam and co-workers[20]proposed three formation pathways for CH2CH2OCH2+(involves AE values of 10.78 eV for channel 2a,11.07 eV for channel 2b and 11.19 eV for channel 2c)and two for CH3OCHCH3+(involves large AE values of 11.91 eV for channel 3a and 11.78 eV for channel 3b)in their detailed G3(MP2) study of the C3H6O+isomers fragmented from DX.Furthermore channel 2b and 2c are suggested to be more efficient in producing CH2CH2OCH2+in their theoretical studies.
From the point of AE value,our experimental AE value of(10.58±0.05)eV suggests perhaps that the channel 2a with a calculated AE value 10.78 eV(also showing good agreement with our theoretical value 10.77 eV)is more favored.Taking into account the facts that C3H6O+involves a number of re-dissociation process[17,21,38],we believe that the three channels mentioned above are possible and may even compete with each other.Particularly,the channel 2a plays a major role in low energies,while the other two channels dominate at high energies.In our study,the β-distonic ion CH2CH2O=CH2+(P3)is assumed to be the only component of the C3H6O+fragment for its decisive abundance.
C.Formation pathway of C3H5O+
As noted by Dunbar and co-workers [38], C3H5O+can begenerated in asequentialway (C4H8O2+→C3H6O++CH2O→C3H5O++H+CH2O)
or a simultaneous way that competes with C3H6O+, C2H5O+and C2H4O+from parent ion.This matches well with our analysis results of relative branching ratios,which suggests that C3H5O+originates from C4H8O2+or C3H6O+or even both. Additionally, the loss of H form C3H6O+must be accompanied by complicated rearrangements and does not produce CH2OCH=CH2+or the oxetanyl cation but dissociates largely via complicated rearrangements yielding CH3CH2CO++H at threshold[17].We thus suppose the sequential channel(CH3CH2CO++CH2O+H)to be the formation pathway of C3H5O+.In the case of CH3CH2CO+(P4a),the first step is 1,4-H shifting to form a relatively stable intermediate INT1 via a five-membered ring transition state TS2 overcoming an energy barrier of 0.97 eV,which is the highest energy step along the entire pathway.The second step is formation of the mentioned important intermediate CH3CH2CHO+via transition state TS3 by prolongation and rotation about the C−O bond.However,the structure of TS3 is not found at B3LYP/6-31+G(d,p) level,but can be found at QCISD/6-31G(d)theoryand MP2 theory[21].The last step is the H atom loss from aldehyde group to form P4a through TS4 by overcoming an energy barrier of 0.75 eV.
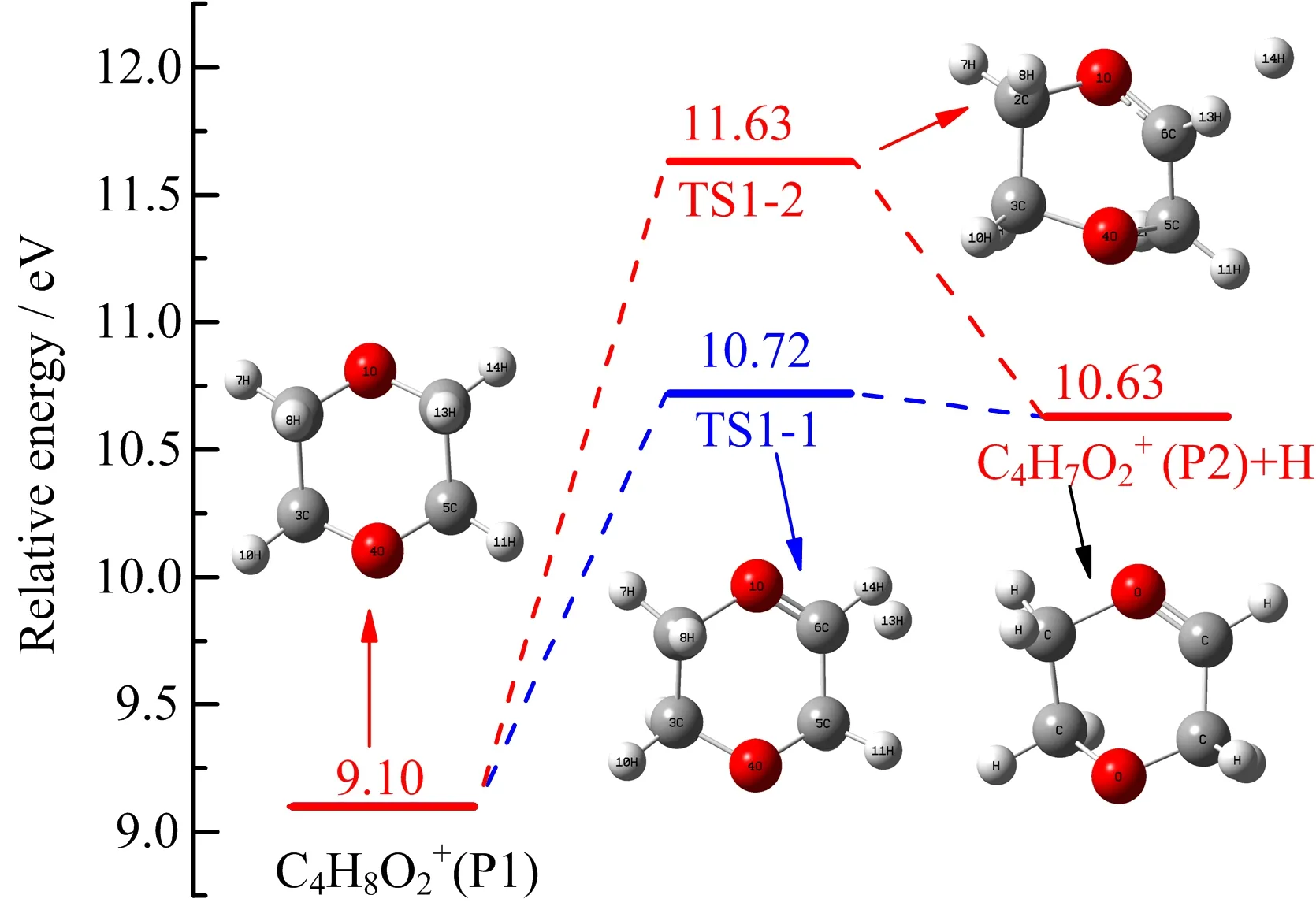
FIG.5 Formation pathway of C4H7O2+calculated at the G3B3//B3LYP/6-31+G(d,p)level.
Obviously,FIG.6(a)shows the sequential fragmentation pathway,while the competitive formation pathway has been shown in FIG.6(b).Considering the facts that the product set CH2OCHCH2++CH2O+H requires3.0eV ofexcitation,whileC3H5O+is observed abundantly at 2.2 eV,the formation of CH2OCHCH2+through this pathway has been precluded. Wethen find acompetitiveformation pathway to produce CH2OCHCH2+(P4b)according to C4H8O2+→CH2OCHCH2+(P4b)+CH2OH,giving a calculated AE value of 11.22 eV closer to experimental one(11.24±0.04)eV.Such plausible mechanism starts from the molecular ion P1 followed by ring-opening and a successive H-shifts to form INT3 via TS5 with an energy barrier of 1.55 eV.CH2OCHCH2+can be formed simply by breaking C2−C3 directly from INT3,which can be further con firmed by scanning the C2−C3 bond length from 1.5˚A to 4.5˚A with a finding that no possible transition state exists.Additionally,the calculated reaction barrier of 2.12 eV is in good agreement with our experimental value of 2.14 eV.We then suppose the latter channel to be the more likely one.
D.Formation pathway of C2H5O+
According to the results of FIG.1 and FIG.2, C2H5O+appears to be one of the dominant as well as the lowest energy fragment ions in the energy region we studied.As for the possible structure of C2H5O+, the first one is CH3OCH2+which is energetic accessible but has been excluded due to its failure to transfer a methyl cation rapidly onto acetone as CH3OCH2+does [13].We also eliminate the structure of protonated oxirane as m/z 45 fragment ion because of a signi ficantly high revised value of the proton affinity while con firm CH3CHOH+as the possible structure of C2H5O+due to its similar enthalpy of formation and speci fical H+transfer rate.The calculated AE of CH3CHOH+is 10.64 eV,agrees reasonably well with our experimental results of(10.52±0.06)eV.
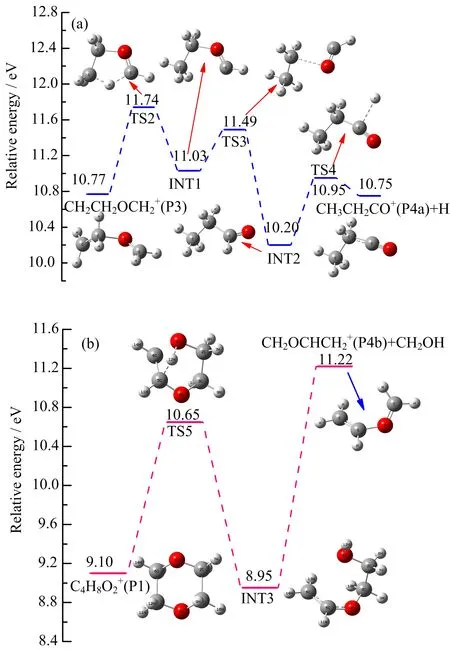
FIG.6Formation pathways of CH3CH2CO+(P4a)and CH2OCHCH2+(P4b)calculated at the G3B3//B3LYP/6-31+G(d,p)level.
Thissen and co-workers[13]proposed an interesting mechanism that the CH3CHOH+can be formed via a H+transfer from the hydroxyl group of CH2CHOH+(a detailed discussion of this charged fragment can be found in the next section)to the aldehyde group of CH3CHO,which gives a rational explanation for the formation of a CH3CHOH+fragment ion,whose AE value is identical to that of the C2H4O+,which is also in good agreement with our experimental results. However,this mechanism cannot be con firmed under the present study.Instead,another formation pathway (P1→TS5→INT3→TS6→INT4)is found.For the purpose of brevity,only the remaining intermediate INT3 and its subsequent steps are discussed.Besides breaking C−C bond to form P4b directly,INT3 can also transform into INT4 via 2,3-H-shift with a transition state TS6 located at 10.23 eV,which is 0.42 eV lower than the highest energy step of TS5. Afterward,a cleavage of C2−O4 bond of INT4 can further dissociate to CH3CHOH+(P5)+CH2CHO.Moreover,if we assume CH3CH2CH2to be the neutral species,the rel-ative energy of COOH++CH3CH2CH2is calculated to be 10.50 eV,which is remarkable close to our experimental AE of(10.52±0.06)eV,but the likelihood of the complex rearrangement as well as the energy barrier required to reach these structures seem uncertain.
E.Formation pathway of C2H4O+
The fragment ions of C2H4O+generated from ionized DX has been reported to possess the enol structure CH2CHOH+but not CH2OCH2+by means of ion-molecule reactions coupling with AE measurements [13].However,the detailed mechanism of this feasible route are still unknown.According to our calculated results in FIG.7 and FIG.8,whether INT3 transforms into TS6 or TS7-2 via 2,3-hydrogen or 2,4-hydrogen shift can lead to different products of similar AE values,indicating that a mechanism via the common intermediate INT3 may account for the same AE values for CH3CHOH+and CH2CHOH+,which has been predicted in the previous discussion section of relative branching ratios. As depicted in FIG.8,the ratelimiting step in CH2CHOH loss is the transfer of the proton,coupled with the cleavage of C2−O5 single bond via the TS7-2(10.76 eV,Table I),the final structure of CH2CHOH++CH2CHOH can be formed,subsequently.
Besides the pathway described above,we have also investigated the possibility of 3,6-hydrogen shift in INT3. On the basis of G3B3 calculation,a channel of little higher AE values(10.99 eV)is found. Staring from INT3,the 3,6-H shift in INT3 leads to the formation of INT5 via TS7-1 with an energy barrier of 1.33 eV.Then,2,3-hydrogen shift and a simple cleavage of C3−O4 bond can transform INT5 into dissociating products CH2CHOH++CH3CHO,overcoming a barrier of 1.57 eV via transition state TS8.The calculated AE 10.76 eV of CH2CHOH+is more close to our experimental AE value(10.52±0.03)eV,suggesting that the dissociative channel CH2CHOH++CH2CHOH is more favored.
F.Formation pathway of C2H3O+
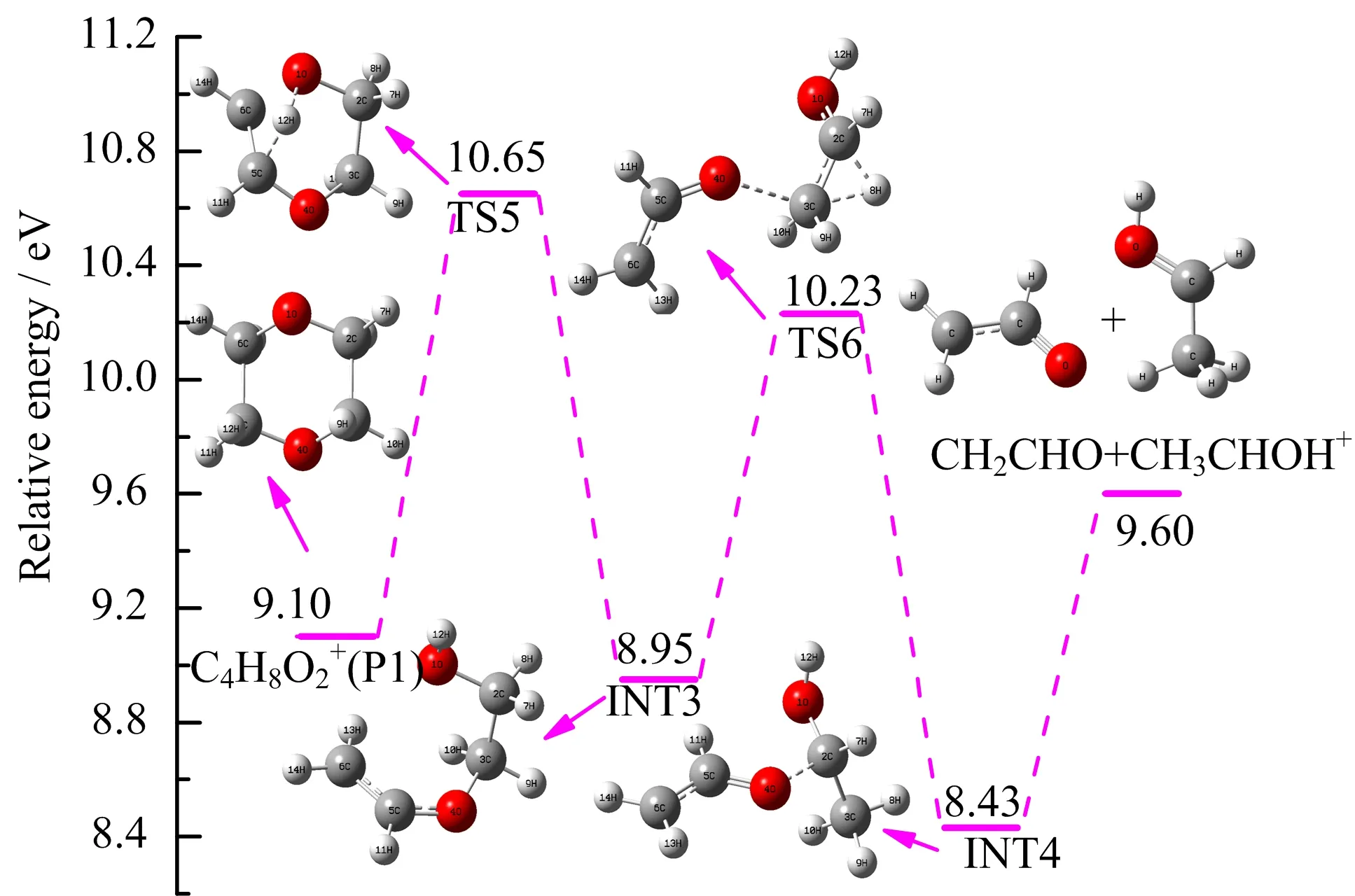
FIG.7 Formation pathways of CH3CHOH+(P5)calculated at the G3B3//B3LYP/6-31+G(d,p)level.
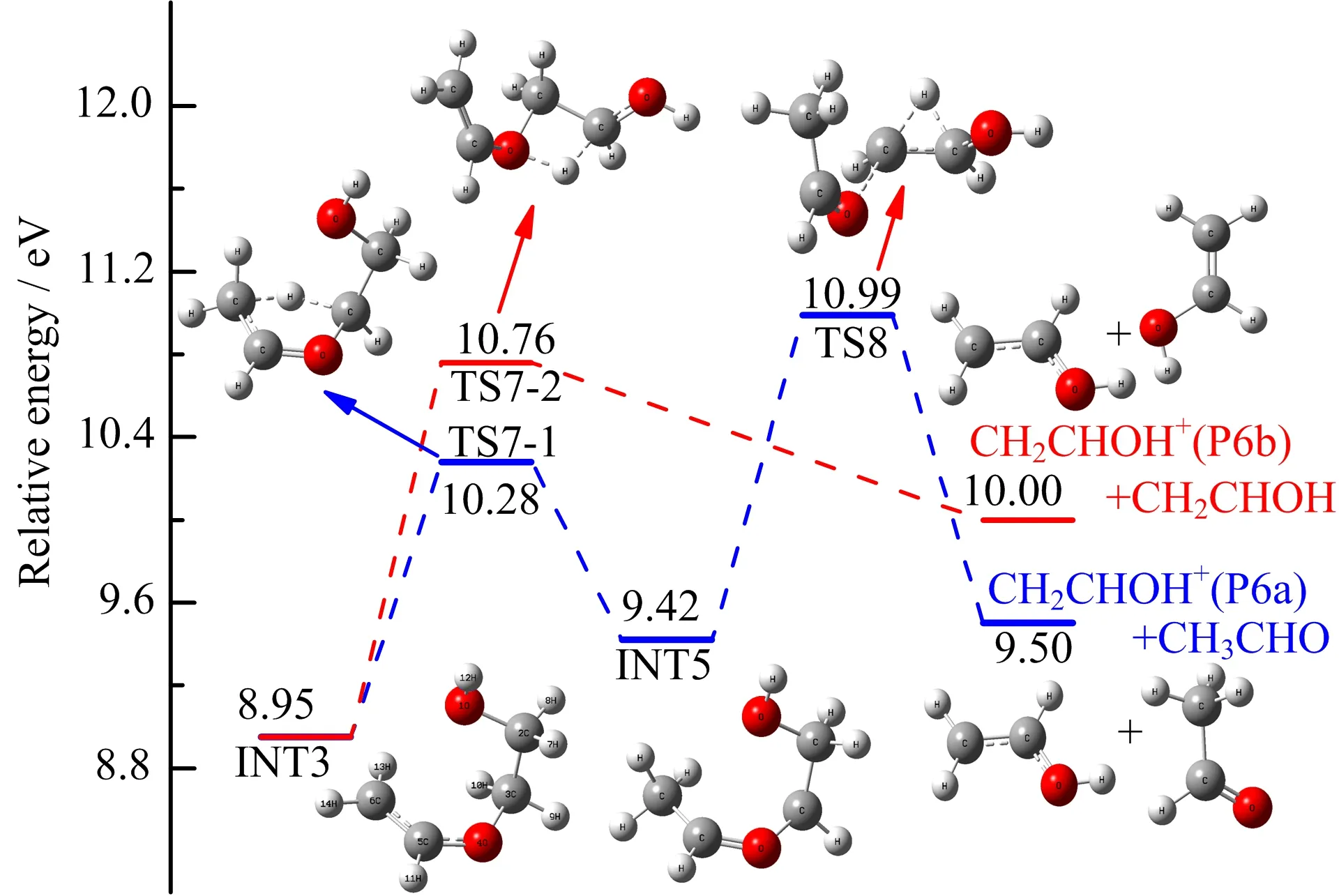
FIG.8 Formation pathways of CH2CHOH+(P6)calculated at the G3B3//B3LYP/6-31+G(d,p)level.
In the case ofC2H3O+, from the analysis of metastable ion spectra of the distonic radical cation CH2CH2OCH2+obtained by neutralizationreionization mass spectrometry(NRMS),Polce et al. [17]have drawn the following three conclusion.Firstly, elimination of CH3to yield C2H3O+can be observed from CH2CH2OCH2+.Secondly,the elimination CH3is exothermic and must,therefore,be associated with sizable activation energies,according to the presence of large kinetic energy releases.Thirdly,the structure of C2H3O+is attributed to be the lowest energy structure of CH3CO+with a calculated standard formation enthalpy value of 799 kJ/mol,which stands much closer to that of C2H3O+(874 kJ/mol)than CH2=C−OH+(784 kJ/mol)favored by Fraser-Monteiro et al.[12]. As shown in Table I,for the CH3CO+,the measured AE value of(12.06±0.10)eV is reasonable consistent with the calculated value of 12.37 eV and is~0.10 eV lower than the experimental value of(11.96±0.10)eV [12].In the present case,a pathway for the formation of CH3CO+is found and the corresponding energy profile is shown in FIG.9.The distonic radical cation (CH2CH2OCH2+,P3)can undergo a hydrogen transfer from the middle C atom to the terminal C atom to generate INT6 via transition state TS9 with an energy barrier of 1.6 eV.It should be noted that INT6 possess the structure of CH3OCHCH2+,which is exactly the mentioned minor component of C3H6O+.This may imply that the two structures of C3H6O+are actually interchangeable.Subsequently,another hydrogen shift between the adjacent C atoms by passing through structure TS10 located at 11.89 eV can explain the formation of INT7 with an elongated C−O bond.Finally,the further cleavage of a weak C−O bond in INT7 can yield CH3and CH3CO+(P7).
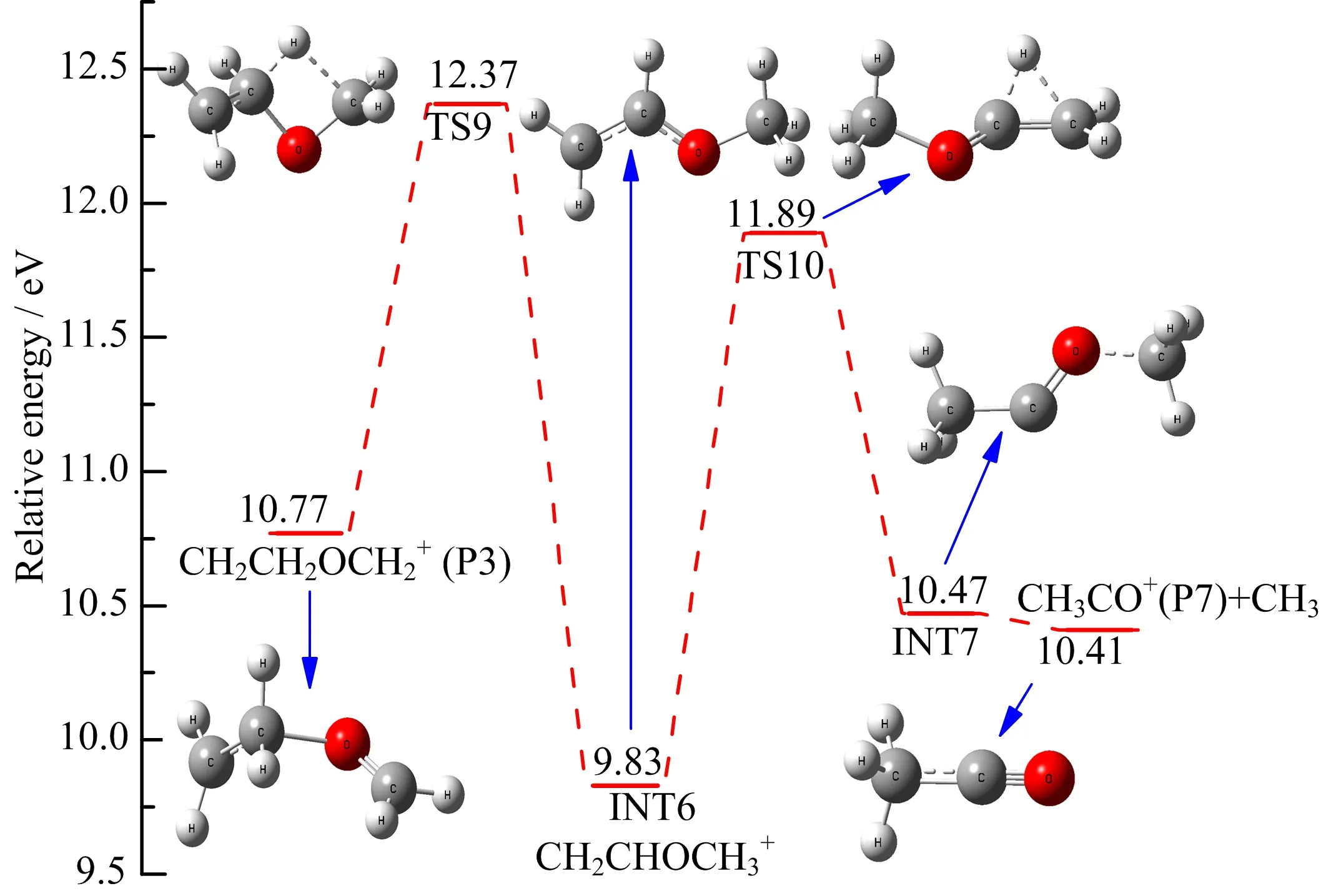
FIG.9 Formation pathways of CH3CO+(P7)calculated at the G3B3//B3LYP/6-31+g(d,p)level.
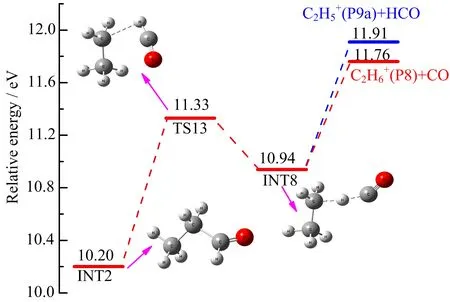
FIG.10 Formation pathways of C2H6+(P8)and C2H5+(P9a)calculated at the G3B3//B3LYP/6-31+g(d,p)level.
G.Formation pathway of C2H6+
Hudson and co-workers[21]explored the formation behavior of C2H6+(m/z 30)and C2H5+(m/z=29)from CH2CH2OCH2+via the pathway: CH2CH2O=CH2+→CH3CH2O=CH+→CH3CH2CHO+→CH3CH2···H···CO+→C2H6++CO or CH3CH2++ HCO by QCISD/6-31G(d)theory forstructures and three given levels of theory for energies. The calculated AE value for C2H6+(P8)is 11.25 eV at QCISD/6-311G(d,p)level,which is somewhat a bit lower than our experimental AE of(11.50±0.03)eV. And in order to compare the energy advantage of the formation pathways of CH3CH2+(P9a)and the CHO+(P9b,will be discussed in the next section), we re-investigate this path at the theoretical level of G3B3//B3LYP/6-31+G(d,p). According to the calculated results,the AE value for C2H6+is 11.76 eV, higher than our experimental AE by 0.26 eV.At this point,the AE values calculated by the mentioned two theoretical methods differ from each other,but are in fair agreement with our experimental AE.The detailed formation pathway of C2H6+is shown in FIG.10. Starting from INT2,a rotation and elongation of C−C bond connecting CH3CH2and CHO leads to the formation of INT8 via transition state TS13 with an energy barrier of 1.13 eV.The calculated structure of INT8 indicates a hydrogen-bridged ion-molecule complex,which is also 0.39 eV lower than the G3B3 energy of TS13(11.33 eV).This may further establish the feasibility of forming INT8.Then the H-bonded complex INT8 can either decompose into C2H6++CO or C2H5++HCO with the H atom immigration.
H.Formation pathway of C2H5+and CHO
A selectively-labelled derivatives study of CH2CH2OCH2+[17]as well as the high-resolution massspectrastudy [12]clearly show thatboth CHO+and C2H5+can be formed competitively. Ourcalculationsindicatethatboth CHO+and C2H5+are energetic possible under G3B3//B3LYP/6-31+G(d,p)level. Then two energetic channels of C4H8O2+→CHO++C2H5+CH2O and C4H8O2+→C2H5++CHO+CH2O are proposed.The AEs value of C2H5+is calculated to be 11.91 eV,showing reasonable agreement with our experimental value of(11.74±0.10)eV. While the calculated AE of CHO+are 12.33 and 12.20 eV,a bit higher than the experimental one,due to the following two formation pathways(shown in FIG.11),respectively.This suggests that C2H5+dominates at lower photon energies, while CHO+appears at higher photon energies.
In the case of CHO+,only the remaining part of pathway proceeding from INT1 is discussed.INT1 undergoes a C−O elongation via transition state TS11 with an energy barrier of 1.17 eV to form the final products CHO+(P9b)+C2H5.Additionally,analogous to the formation of CH3CH2CO+(P4a),CHO+(P9b)can also be formed through INT2 in which the CH3CH2−CHO bond is elongated with a relative high energy barrier of 2.13 eV.Subsequently,dissociation of TS12 yields C2H5+CHO+as products.
I.Formation pathway of C2H4+
A typical structure of CO+,with the same m/z=28 as C2H4+,has been excluded by previous high-resolution mass spectra[12]and a metastable ion mass spectra study[17]of deuterated C3D6O+,where the original mass peak of m/z=28 shifts to 32. Therefore,the m/z=28 species is assigned to C2H4+. The formation of C2H4+from that one step dissociation channels C2H4++OCH2CH2O and C2H4++c-OCH2CH2O(four-membered-ring)would require energy of at least 15.47 and 14.47 eV,respectively, which are much higher than our experimental result(11.78±0.10)eV as well as the literature valuesof(11.90±0.10)eV[12]and 11.8 eV[15]. Whereas, the formation process simply by breaking bond can be clearly eliminated. From FIG.2,the branching ratio of C2H4+increases with the decreasing of the branching ratio of C3H6O+with energy increasing,suggesting that C2H4+is formed by C3H6O+. Then a higher energy consecutive dissociation path C4H8O2+→C3H6O++CH2O→C2H4++2CH2O is proposed.As shown in FIG.11,the calculated AE value of this consecutive dissociation path is 12.02 eV,showing reasonable agreement with experimental result of (11.78±0.10)eV.
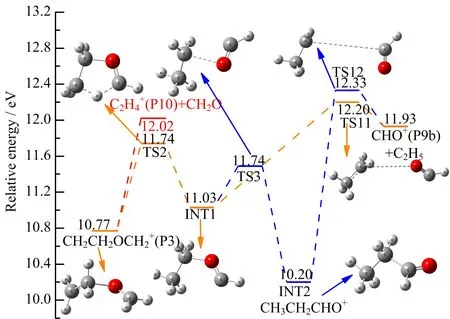
FIG.11 Formation pathways of CHO+(P9a)and C2H4+(P10)calculated at the G3B3//B3LYP/6-31+G(d,p)level. The energy of neutral 1,4-dioxane is de fined as zero.
IV.CONCLUSION
Dissociative photoionization of DX was investigated with VUV-TOF-PIMS in conjunction with supersonic expansion molecular beam.IE of parent molecule,AE values of twelve observed fragment ions,and relative branching ratios of major ions(m/z=88,87,58,57,45, 44,43,31,30,29,and 28)were derived from their PIE curves.A few of the determined values are revised with respect to previous publications.Based on comparison of the experimental and theoretical AE values,detailed dissociative photoionization channels of R1−R11 are identi fied R1:C4H7O2+(P2,m/z=87)+H,
R2:CH3CH2CO+(P4a,m/z=57)+H+CH2O,
R3:CH2CHOCH2+(P4b,m/z=57)+H+CH2O,
R4:CH3CHOH+(P5,m/z=45)+CH2CHO,
R5:CH2CHOH+(P6,m/z=44)+CH3CHO,
R6:CH2CHOH+(P6,m/z=44)+CH2CHOH,
R7:CH3CO+(P7,m/z=43)+CH3+CH2O,
R8:C2H6+(P8,m/z=30)+CO+CH2O,
R9:C2H5+(P8a,m/z=29)+CHO+CH2O,
R10:CHO+(P8b,m/z=29)+C2H5+CH2O
R11:C2H4+(P9,m/z=28)+CH2O+CH2O
C3H5O+isreferred tobeCH3CH2CO+and CH2CHOCH+2respectively. They can be generated by further elimination ofH from the CHO group of CH3CH2CHO+and elimination of CH2OH from the molecular ion. Fragment ions with m/z=29 are attributed to C2H5+andCHO+whichcanbeproducedvia CH2CH2O=CH2+→CH3CH2O=CH+→CH3CH2CHO+→CH3CH2···H···CO+→C2H5++HCO and by elimination ofC2H5through CH3CH2OCH+or CH3CH2CHO+or even both respectively.The present study is of great signi ficance for understanding the photoionization and dissociation processes of DX in the photon energy region of 8.00−15.50 eV.
V.ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China(No.91544105,No.U1532137, No.11575178,and No.U1232209).
[1]D.K.Stepien,P.Diehl,J.Helm,A.Thoms,and W. Puttmann,Water Res.48,406(2014).
[2]M.J.Zenker,R.C.Borden,and M.A.Barlaz,Environ. Eng.Sci.20,423(2003).
[3]D.T.Adamson,S.Mahendra,K.Walker,S.Rauch,S. Sengupta,and C.Newell,Environ.Sci.Technol.Lett. 1,254(2014).
[4]C.T.Derosa,S.Wilbur,J.Holler,P.Richter,and Y. W.Stevens,Toxicol.Ind.Health 12,1(1996).
[5]J.Y.Choi,Y.J.Lee,J.Shin,and J.W.Yang,J.Hazard.Mater.179,762(2010).
[6]W.Shen,H.Chen,and S.Pan,Bioresour.Technol.99, 2483(2008).
[7]M.J.Zenker,R.C.Borden,and M.A.Barlaz,J.Environ.Eng.130,926(2004).
[8]J.Platz,J.Sehested,O.J.Nielsen,and T.J.Wallington,J.Chem.Soc.Faraday Trans.93,2855(1997).
[9]T.Maurer,H.Hass,I.Barnes,and K.H.Becker,J. Phys.Chem.A 103,5032(1999).
[10]J.E.Collin and G.Cond´e,Bull.Class Sci.Acad.Roy. Belg.52,978(1966).
[11]J.C.Traeger,Org.Mass Spectrom.20,223(1985).
[12]M.L.Fraser-Monteiro,J.J.Butler,and T.Baer,J. Phys.Chem.86,739(1982).
[13]R.Thissen,P.Mourgues,and H.E.Audier,Eur.Mass Spectrom.4,79(1998).
[14]R.Thissen,H.E.Audier,J.C.Rooke,and P.Mourgues,Eur.Mass Spectrom.5,147(1999).
[15]P.Zou,G.S.Wu,W.W.Chen,D.L.Yang,L.S.Sheng, G.H.Wu,W.Q.Ye,and Y.W.Zhang,J.Acta.Phys-Chim.14,21(1998).
[16]J.Jalonen,J.M.Tedder,and P.H.Nidaud,J.Chem. Soc.Faraday Trans.II 76,1450(1980).
[17]M.J.Polce and C.Wesdemiotis,J.Am.Chem.Soc. 115,10849(1993).
[18]A.B.Burrill and P.M.Johnson,Chem.Phys.Lett. 350,473(2001).
[19]K.Watanabe,T.Nakayama,and J.Mottl,J.Quant. Spectry.Radiative Transfer.2,369(1962).
[20]C.S.Lam,W.K.Li,and S.W.Chiu,J.Phys.Chem. A 109,7296(2005).
[21]C.E.Hudson,D.J.McAdoo,and J.C.Traeger,J.Am. Soc.Mass Spectrom.13,1235(2002).
[22]J.Chen,M.Q.Cao,B.Wei,M.M.Ding,X.B.Shan, F.Y.Liu,and L.S.Sheng,J.Mass Spectrom.51,169 (2016)
[23]M.Q.Cao,Y.Q.Li,G.B.Chu,J.Chen,X.B.Shan, F.Y.Liu,Z.Y Wang,and L.S.Sheng,J.Electron Spectrosc.Relat.Phenom.191,41(2013).
[24]W.Z.Fang,L.Gong,Q.Zhang,X.B.Shan,F.Y.Liu, and L.S.Sheng,J.Chem.Phys.134,174306(2011).
[25]W.X.Li,Y.J.Hu,J.W.Guan,F.Y.Liu,X.B.Shan, and L.S.Sheng,J.Chem.Phys.139,024307(2013).
[26]B.Franziska,N.R.Qiao,G.Amir,Paul R.Horn,A. Musahid,R.L.Stephen,and H.G.Martin,J.Am. Chem.Soc.135,14229(2013).
[27]M.C.Castrovilli,P.Bolognesi,A.Cartoni,D.Catone, P.O’Keeffe,A.R.Casavola,S.Turchini,N.Zema,and L.Avaldi,J.Am.Soc.Mass Spectrom.25,351(2014).
[28]X.F.Tang,X.G.Zhou,M.Niu,S.Liu,J.Sun,X.B. Shan,F.Y.Liu,and L.S.Sheng,Rev.Sci.Instrum. 80,113101(2009).
[29]S.Zhang,Y.M.Wang,Z.Z.Cao,B.Zhang,S.S.Wang, R.H.Kong,Y.J.Zhao,X.B.Shan,and L.S.Sheng, Rev.Sci.Inst.78,043104(2007).
[30]R.H.Kong,X.B.Shan,S.S.Wang,Y.W.Zhang, L.S.Sheng,L.Q.Hao,and Z.Y.Wang,J.Electron Spectrosc.Relat.Phenom.160,49(2007).
[31]X.Y.Liu,W.J.Zhang,Z.Y.Wang,M.Q.Huang, X.B.Yang,L.Tao,Y.Sun,Y.T.Xu,X.B.Shan,F. Y.Liu,and L.S.Sheng,J.Mass.Spectrom.44,404 (2009).
[32]W.Z.Fang,G.Lei,X.B.Shan,F.Y.Liu,Z.Y.Wang, and L.S.Sheng,J.Electron Spectrosc.Relat.Phenom. 184,129(2011).
[33]W.Z.Fang,L.Gong,X.B.Shan,Y.J.Zhao,F.Y. Liu,Z.Y.Wang,and L.S.Sheng,J.Mass.Spectrom. 46,1152(2011).
[34]W.Z.Fang,L.Gong,X.B.Shan,F.Y.Liu,Z.Y. Wang,and L.S.Sheng,Anal.Chem.83,9024(2011).
[35]M.J.Frisch,G.W.Trucks,H.B.Schlegel,G.E. Scuseria,M.A.Robb,J.R.Cheeseman,G.Scalmani, V.Barone,B.Mennucci,G.A.Petersson,H.Nakatsuji,M.Caricato,X.Li,H.P.Hratchian,A.F.Izmaylov,J.Bloino,G.Zheng,J.L.Sonnenberg,H. Hada,M.Ehara,K.Toyota,R.Fukuda,J.Hasegawa, M.Ishida,T.Nakajima,Y.Honda,O.Kitao,H.Nakai, T.Vreven,J.A.Montgomery,Jr.,J.E.Peralta,F. Ogliaro,M.Bearpark,J.J.Heyd,E.Brothers,K.N. Kudin,V.N.Staroverov,R.Kobayashi,J.Normand, K.Raghavachari,A.Rendell,J.C.Burant,S.S.Iyengar,J.Tomasi,M.Cossi,N.Rega,J.M.Millam,M. Klene,J.E.Knox,J.B.Cross,V.Bakken,C.Adamo, J.Jaramillo,R.Gomperts,R.E.Stratmann,O.Yazyev, A.J.Austin,R.Cammi,C.Pomelli,J.W.Ochterski, R.M.Martain,K.Morokuma,V.G.Zakrzewski,G.A. Voth,P.Salvador,J.J.Dannenberg,S.Dapprich,A. D.Daniels,O.Farkas,J.B.Foresman,J.V.Ortiz,J. Cioslowski,and D.J.Fox,Gaussian 09,Revision A.1, Wallingford,CT:Gaussian,Inc.,(2009).
[36]http://webbook.nist.gov/Last accessed(2017).
[37]R.Wada and M.Kato,Chem.Phys.Lett.641,74 (2015).
[38]R.C.Dunbar,F.S.Huang,and S.J.Klippenstein,Int. J.Mass Spectrom.Ion Proc.128,2l(1993).
∗Author to whom correspondence should be addressed. E-mail:fyliu@ustc.edu.cn,Tel:+86-551-63602123,FAX:+86-551-65141078
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- γ-Ray Irradiation-Derived MnO/rGO Composites for High Performance Lithium Ion Batteries
- Identi fication of Superoxide O2−during Thermal Decomposition of Molten KNO3-NaNO2-NaNO3Salt by Electron Paramagnetic Resonance and UV-Vis Absorption Spectroscopy
- Binding Mechanism and Molecular Design of Benzimidazole/Benzothiazole Derivatives as Potent Abl T315I Mutant Inhibitors
- Highly Responsive and Selective Ethanol Gas Sensor Based on Co3O4-Modi fied SnO2Nano fibers
- Geometric Design of Anode-Supported Micro-Tubular Solid Oxide Fuel Cells by Multiphysics Simulations
- Laser-Assisted Stark Deceleration of Polar Molecules HC2n+1N(n=2,3,4) in High-Field-Seeking State