脑脊液MOG抗体阳性的视神经脊髓炎谱系疾病患者的临床特征
2017-09-03李昕頔王化冰周衡周安娜刘永红赵琳张星虎
李昕頔 王化冰 周衡 周安娜 刘永红 赵琳 张星虎
脑脊液MOG抗体阳性的视神经脊髓炎谱系疾病患者的临床特征
李昕頔 王化冰 周衡 周安娜 刘永红 赵琳 张星虎
目的 探讨MOG抗体阳性的NMOSD患者的临床特点。方法 选择29例NMOSD 患者,根据血清AQP-4 抗体以及脑脊液MOG抗体检测结果,分为MOG抗体阳性、AQP4抗体阳性的NMOSD(剔除双阳性者),同时选择13例MS患者作为对照。回顾性分析上述三组患者临床信息,统计归纳其临床特点。结果 29例NMOSD患者中血清AQP4抗体阳性者11例,脑脊液MOG抗体阳性者8例。36.4%(4例/11例)AQP4抗体阳性、62.5%(5例/8例)MOG抗体阳性NMOSD患者,以及7.7%(1例/13例)MS患者合并脊髓炎与视神经炎,三组间差异有统计学意义(χ2=7.128,P=0.028),其中MOG抗体阳性NMOSD患者较MS患者更易合并视神经炎(χ2=7.289,P=0.014)。MOG抗体阳性NMOSD患者缓解期EDSS分数低于AQP4抗体阳性NMOSD患者〔3.50 (2.50, 4.00),4.00 (3.50, 6.00) ,Z=-2.379,P=0.020〕。MOG抗体阳性NMOSD脊髓病灶多表现为多发的长节段脊髓病灶,50%(4例/8例)MOG抗体阳性脊髓病灶个数大于1个,与MS组无明显差异,而AQP4抗体阳性组均为单个病灶。MOG抗体阳性NMOSD脊髓病灶长度较AQP4抗体阳性组短〔分别(3(2,3)个椎体、4(3,5)个椎体,Z=-2.499,P=0.012〕,较MS组〔(1.25(1,1.5)个椎体〕长(Z=-3.447,P<0.001)。8例MOG抗体阳性患者中5例存在颅内病灶,3例表现为NMOSD样颅内病灶,余2例表现为MS样颅内病灶,其病灶形态及部位与AQP4抗体阳性组无明显差异,而与MS组存在差异。结论 MOG抗体阳性NMOSD合并视神经炎的患者较多,临床残障程度较轻,预后较好,脊髓病灶为多发的长节段脊髓病灶;颅内病灶的形态及部位与MS无明显差异。
视神经脊髓炎谱系疾病;多发性硬化;水通道蛋白4抗体;髓鞘少突胶质细胞糖蛋白抗体;磁共振波谱学
视神经脊髓炎(Neuromyelitis optica,NMO)是一种自身免疫相关的中枢神经系统炎性脱髓鞘疾病,患者主要表现为复发性长节段横贯性脊髓炎和(或)视神经炎。血清或脑脊液水通道蛋白4 (aquaporin-4,AQP-4)抗体在 NMO 诊断中具有高度特异性[1],该抗体在鉴别NMO与MS中十分重要。2007年视神经脊髓炎谱系疾病(neuromyelitis optica spectrum disorder,NMOSD)的概念被提出,其中囊括了AQP4抗体阴性、伴脑内非典型病灶的非典型NMO患者[2]。2015年国际NMO诊断小组修订了NMOSD的诊断标准,将既往经典的NMO归为NMOSD的一部分,该诊断标准将NMOSD分为AQP4抗体阳性和AQP4抗体阴性两大类,扩大既往NMSOD的疾病谱[3]。
髓鞘少突胶质细胞糖蛋白(myelin oligodendrocyte glycoprotein,MOG)是中枢神经系统(central nervous system,CNS)髓鞘最外层含量极微的髓鞘蛋白成分。MOG抗体是免疫球蛋白IgG1的一种亚型,可以有效地调节补体依赖性细胞毒性反应。既往研究者认为MOG抗体在MS的发病中发挥了重要的作用[4]。随后,有研究者在部分AQP4抗体阴性的NMOSD患者血清中发现了MOG抗体,并发现其临床特点与经典AQP4抗体阳性的NMOSD有所区别,认为二者发病机制可能不同[5],由MOG抗体介导的免疫性疾病可能是一种不同于AQP4抗体阳性的NMOSD或MS的疾病。目前关于MOG抗体阳性的NMOSD患者临床特点的国内研究较少。本研究回顾性分析比较脑脊液MOG抗体阳性NMOSD与经典AQP4抗体阳性NMOSD以及MS患者之间临床及影像学特征,进一步探究中国MOG抗体阳性的NMOSD患者的临床特点,期望有助于今后对国内NMOSD患者的诊断与治疗。
1 对象和方法
1.1 观察对象 自2015-06—2016-06期间就诊并随诊于北京天坛医院神经感染免疫科的中枢系统炎性脱髓鞘患者42例,包括:(1)NMOSD患者29例,男7例,女21例;年龄为15~71岁,中位年龄(上、下四分位数)为44(27,55)岁;发病年龄为13~70岁,中位发病年龄(上、下四分位数)为38(24,46)岁;病程为11~146个月,中位病程(上、下四分位数)为47(32,83)个月。所有NMOSD患者均符合2015年国际 NMO 诊断小组修订的 NMOSD诊断标准[3],并且急性期均未接受大剂量糖皮质激素或免疫抑制剂治疗时采取血清与脑脊液标本。(2)MS患者13例,男4例,女9例;年龄为16~63岁,中位年龄(上、下四分位数)为43(24,47)岁;发病年龄为15~63岁,中位发病年龄(上、下四分位数)为30(20,42)岁;病程(上、下四分位数)为8~192个月,中位病程(上、下四分位数)为18(12,90)个月。患者符合2010年McDonald MS诊断标准[6]。
所有患者均不存在其他严重内科疾病,能配合研究。
1.2 方法 回顾性分析29例NMOSD 患者及13例MS患者的临床资料,包括发病年龄、性别、病程、临床症状、临床残障程度以及磁共振(magnetic resonance imaging,MRI)表现等。应用免疫荧光整细胞法[7](cell-based assay,CBA)定性检测NMOSD患者的血清 AQP-4 抗体;采用CUSABIO公司的人抗髓鞘少突胶质细胞糖蛋白抗体(IgG)酶联免疫试剂盒,经酶联免疫吸附试验(enzyme linked immunosorbent assay,ELISA)定性检测脑脊液MOG抗体。
所有患者均于急性期,症状最重时行急性期扩展的功能障碍状况量表(expanded disability status scale,EDSS)评分,经大剂量甲泼尼龙冲击治疗后(起始剂量为1000 mg或500 mg)行缓解期EDSS评分。
根据血清AQP4抗体及脑脊液MOG抗体检测结果,在排除双阴性及双阳性NMOSD患者后,将NMOSD患者分为AQP4抗体阳性和MOG抗体阳性两组,以AQP4抗体阳性组及MS患者为对照组,对比分析三组患者的临床资料。

2 结果
2.1 基本信息及临床表现 29例为NMOSD患者中,血清AQP4抗体阳性者11例,脑脊液MOG抗体阳性者8例,双阴性者10例,无双阳性者。MOG抗体阳性、AQP4抗体阳性NMOSD患者以及MS患者均为女性多见,三组间差异无统计学意义。三组间发病年龄、病程差异均无统计学意义。具体见表1。
19例NMOSD患者均存在急性脊髓炎症状, MS患者仅有7例(53.8%)存在急性脊髓炎症状,两者差异有统计学意义(P= 0.02)。 脊髓炎的临床表现主要包括肢体肌力下降、肢体麻木、痛温觉及震动觉等感觉功能下降,以及二便功能障碍等症状。其中62.5%(5例/8例)MOG抗体阳性NMOSD患者、36.4%(4例/11例)AQP4抗体阳性NMOSD患者以及7.7%(1例/13例)MS患者同时合并脊髓炎与视神经炎,三组间差异有统计学意义(P=0.028)。其中MOG抗体阳性NMOSD患者较MS患者更易合并视神经炎(P=0.014),但抗体阳性的两NMOSD组间差异无统计学意义。具体见表2。
2.2 急性期及缓解期临床神经功能残障程度 三组间急性期EDSS评分差异无统计学意义(P=0.057)。MOG抗体阳性NMOSD患者缓解期EDSS评分及EDSS评分差值均低于AQP4抗体阳性NMOSD患者(P<0.05),而均与MS患者比较差异无统计学意义(P≥0.50)。具体见表3。
2.3 影像学表现
2.3.1 脊髓MRI表现:具体见表4、图1-3。19例NMOSD患者均存在脊髓病灶,仅有7例(53.8%)MS患者存在脊髓病灶。其中MOG抗体阳性NMOSD患者比MS患者更易出现脊髓病灶(P=0.046),与AQP4抗体阳性NMOSD患者间差异无统计学意义。MOG抗体阳性NMOSD患者中,4例病灶位于颈髓,5例病灶位于胸髓。三组的脊髓病灶数量存在差异,MOG抗体阳性NMOSD患者的脊髓病灶数量较AQP4抗体阳性NMOSD患者多(P=0.018),与MS患者间差异无统计学意义。三组患者的脊髓病灶节段数也存在差异,MOG抗体阳性组脊髓病灶所对应的中位椎体个数比AQP4抗体阳性NMOSD患者少(P=0.012),比MS患者多(P<0.01)。
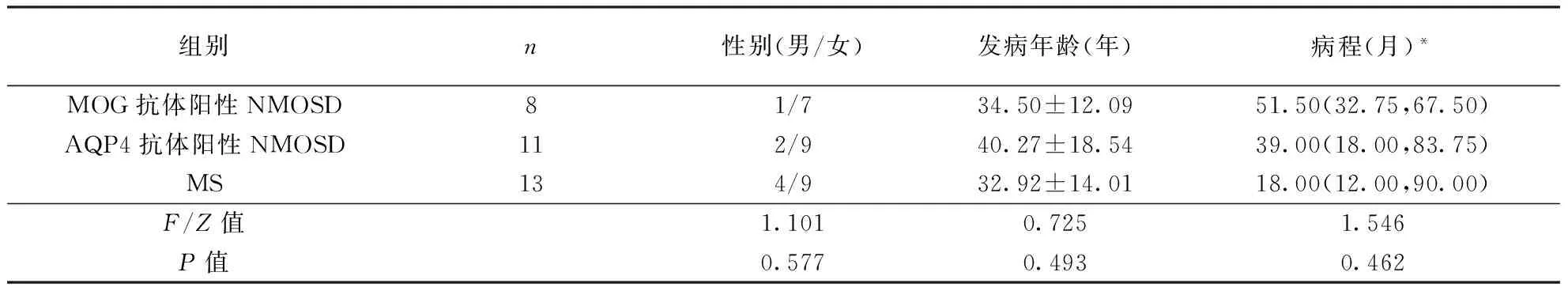
表1 各组患者临床基本信息的比较
注:*中位数(上、下四分位数)

表2 各组患者临床症状的比较〔n(%)〕


表3 各组急性期及缓解期EDSS评分比较〔M(P25,P75)〕


表4 三组患者脊髓MRI的特点
注释:P1值为MOG抗体阳性NMOSD患者、AQP4抗体阳性NMOSD患者两组间比较所得;P2值为MOG抗体阳性NMOSD患者、MS患者两组间比较所得

图1 1例MOG抗体阳性的NMOSD病患者颈髓病灶表现:第6-7胸椎、第11胸椎-第1腰椎脊髓内可见片状长T2信号 图2 1例AQP4抗体阳性NMOSD患者的颈髓病灶表现:第2-7颈髓内可见长T2信号 图3 1例MS患者的颈髓病灶表现:第3-5颈髓内可见不连续的短片状长T2信号
2.3.2 头颅MRI表现:具体见表5、图4、5。11例AQP4抗体阳性NMOSD患者中9例存在颅内病灶,其中5例患者表现为典型的NMOSD颅内病灶(图4);4例患者表现为侧脑室旁或大脑皮层下非特异性斑点状病灶。8例MOG抗体阳性患者中5例存在颅内病灶,病灶部位及形态具有多样性(图5),其中2例颅内病灶分布于在第四脑室周围室管膜区(即桥臂及延髓上段病灶),1例病灶位于右侧侧脑室旁,表现为大脑半球广泛融合性病灶(最大直径约为3 cm),与AQP4抗体阳性NMOSD患者的颅内病灶位置形态相似;另外2例表现为位于底节区及侧脑旁,类似MS样病灶。MOG抗体阳性NMOSD患者颅内病灶较MS患者多表现为典型的NMOSD样病灶,即位于室管膜周围或大脑半球广泛融合性病灶。MOG抗体阳性NMOSD患者与AQP4抗体阳性NMOSD患者相比,病灶形态、部位无明显差异。

表5 三组患者颅内MRI病灶的特点比较〔n(%)〕
注:P1值为MOG抗体阳性NMOSD患者、AQP4抗体阳性NMOSD患者两组间比较所得;P2值为MOG抗体阳性NMOSD患者、MS患者两组间比较所得。典型NMOSD颅内病灶主要指位于室管膜周围或皮层下广泛融合性病灶;MS样病灶主要指位于侧脑室旁多发的类椭圆状病灶,长轴常垂直于侧脑室

图4 AQP4抗体阳性组中的典型NMOSD病灶表现:右侧四脑室旁桥臂处大片状异常FLAIR信号(A),颈髓与延髓交界处长T2信号(B),左侧额叶皮层及皮层下白质大片状异常FLAIR信号(C) 图5 MOG抗体阳性组中3例患者的典型NMOSD病灶表现:右侧四脑室旁桥臂处小片状长T2信号(A),延髓类圆形及线状长T2信号(B),右侧脑室旁丘脑及底节区可见片状异常FLAIR信号(C)
3 讨论
伴随着AQP4抗体被逐渐认识,NMO已成为一种独立的自身免疫性中枢神经系统炎性脱髓鞘疾病。研究表明50%以上的NMOSD患者血清AQP4抗体阳性。近几年一项包含215例NMOSD患者的研究示约有21%的AQP4 抗体阴性NMOSD患者的血清MOG抗体高表达[8],这类患者临床特征与AQP4抗体阳性患者有所差异。本研究中应用ELISA方法测定MOG抗体,该方法采用线性的 MOG 蛋白碎片,存在非特异性抗体的吸附,其特异度不如CBA方法。本研究为保证检测准确性,一方面检测脑脊液MOG抗体而非血清抗体,另一方面MOG试剂盒本身存在阴性对照与阳性对照来确保检测的灵敏度与特异度。
国外一项包含100例NMOSD患者研究发现约有40% MOG抗体阳性患者同时出现视神经炎与脊髓炎,并且明显高于AQP4抗体阳性组,该文献作者推断MOG抗体更易造成视神经的损伤[5]。另有研究发现MOG抗体阳性患者视神经乳头肿胀往往较AQP4抗体阳性患者更为明显[9]。本项研究发现MOG抗体阳性的NMOSD患者同时合并视神经炎与急性脊髓炎的例数较MS患者多,而与AQP4抗体阳性的NMOSD患者间差异无统计学意义,提示MOG抗体阳性患者的视神经较MS患者更易受累。
目前临床研究中常用EDSS评价NMOSD患者的临床神经功能缺损程度。有研究报道MOG抗体阳性的NMOSD患者较AQP4抗体阳性患者的急性期及缓解期EDSS分数降低[10]。本研究结果与上述结果相似,MOG抗体阳性NMOSD患者的急性期及缓解期EDSS分数较AQP4抗体阳性患者低,较MS患者高。经急性期大剂量激素冲击治疗后,MOG抗体阳性患者缓解期与急性期EDSS分数差值明显大于AQP4抗体阳性患者(P=0.033)。上述现象可能与AQP4抗体和MOG抗体引起的病理改变不同有关。有研究发现将 MOG 抗体注射到小鼠大脑内,髓鞘细胞表面蛋白的表达发生改变,引起非补体依赖性炎性反应,无神经元坏死或大量炎性细胞浸润,病变可在2 周内自行修复[11];而AQP4抗体往往引起瀑布性炎性反应,病灶内可见神经元及星形胶质细胞不可逆坏死,周围大量中性粒细胞、嗜酸性粒细胞浸润[12]。因此临床上观察到MOG抗体阳性NMOSD患者残障程度较AQP4抗体阳性患者轻,且经治疗后残障程度恢复明显,预后较好。目前MOG抗体具体致病机制尚不明确。
NMOSD脊髓病灶多位于颈、胸髓内,长度大于3个连续椎体数[2]。一项包含50例MOG抗体阳性患者的研究中发现82.1% 的病灶位于颈髓,75%位于胸髓[13],而本研究中MOG抗体阳性患者的胸髓病灶略多于颈髓病灶。与一项纳入93例NMOSD的研究结果相似[14],本研究发现多数MOG抗体阳性NMOSD患者为多发的脊髓病灶,与MS类似,而AQP4抗体阳性组多为单发的脊髓病灶。MOG抗体阳性NMOSD脊髓病灶长度为2~5个椎体节段[13]。本研究中MOG抗体阳性组的中位脊髓病灶长为3个椎体节段,较AQP4抗体阳性组短,较MS脊髓病灶长,与既往文献结果相似。综上可见,MOG抗体阳性NMOSD的脊髓病灶特点为:常见于颈胸髓内,多表现为多发的长节段脊髓病灶,但其长度短于AQP4抗体阳性的NMOSD患者。
NMOSD典型颅内病灶多位于AQP4高表达的脑室系统周围室管膜区[15]。目前2项国内外相关队列研究均表示在MOG抗体阳性的NMOSD患者中未见到典型性的NMOSD头部病灶,其头部病灶多类似于MS或急性播散性脑脊髓膜炎(ADEM)样改变,或者表现为颅内非特异性点状病灶[5, 14]。个别病例报道称MOG抗体阳性NMOSD患者也可出现典型的NMOSD颅内病变,即累及第三脑室及中脑导水管周围的间脑,包括丘脑、下丘脑和中脑后缘;累及延髓出现类似线状延续性颈髓病灶;或累及大脑半球白质广泛融合肿瘤样病灶,一般无占位效应[16]。本研究发现8例MOG抗体阳性患者中仅2例出现典型MS样病灶,而3例患者出现位于第四脑室周围室管膜区或皮层下的片状典型NMOSD病变。与既往研究有所不同,本研究发现MOG抗体阳性的NMOSD患者并非多表现为MS样或ADEM样颅内病灶,一部分患者也可表现为典型的NMOSD样病变。因此我们认为MOG抗体阳性NMOSD患者的颅内病灶的形态与部位无明显特异性。
本研究存在一定的局限性。首先纳入样本量较小;其次,本研究MOG抗体检测方法为经典ELISA法,其特异性及灵敏度低于CBA法,存在假阳性与假阴性的可能性。在未来研究中可以采用细胞为基础的检测方法,设立阴性对照,扩大样本量,进一步探讨MOG抗体阳性NMOSD患者的临床特点。
综上所述,本研究发现MOG抗体阳性的NMOSD患者易合并视神经炎,脊髓病灶为多发的长节段脊髓病灶,颅内病灶的形态与部位与AQP4抗体阳性NMOSD患者相比无明显特异性,疗效及预后均好于AQP4抗体阳性的NMOSD。因此,对于临床上高度怀疑为NMOSD但AQP4抗体为阴性的脱髓鞘病患者,可以完善血清或脑脊液MOG抗体检测,便于早期明确诊断治疗,改善患者预后。
[1]Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis[J]. Lancet, 2004, 364(9451):2106-2112.
[2]Wingerchuk DM, Lennon VA, Lucchinetti CF, et al. The spectrum of neuromyelitis optica[J]. Lancet Neurol, 2007, 6(9):805-815.
[3]Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders[J]. Neurology, 2015, 85(2):177-189.
[4]Zadro I, Brinar V, Horvat G, et al. Clinical relevance of antibodies against myelin oligodendrocyte glycoprotein in different clinical types of multiple sclerosis[J]. Clin Neurol Neurosurg, 2007, 109(1):23-26.
[5]van Pelt ED, Wong YY, Ketelslegers IA, et al. Neuromyelitis optica spectrum disorders: comparison of clinical and magnetic resonance imaging characteristics of AQP4-IgG versus MOG-IgG seropositive cases in the Netherlands[J]. Eur J Neurol, 2016, 23(3):580-587.
[6]Yamout B, Alroughani R, Al-Jumah M, et al. Consensus guidelines for the diagnosis and treatment of multiple sclerosis[J]. Curr Med Res Opin, 2013, 29(6):611-621.
[7]Yang CS, Zhang DQ, Wang JH, et al. Clinical features and sera anti-aquaporin 4 antibody positivity in patients with demyelinating disorders of the central nervous system from Tianjin, China[J]. CNS Neurosci Ther,2014, 20(1):32-39.
[8]Sato DK, Callegaro D, Lana-Peixoto MA, et al: Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders[J]. Neurology, 2014, 82(6):474-481.
[9]Ramanathan S, Reddel SW, Henderson A, et al: Antibodies to myelin oligodendrocyte glycoprotein in bilateral and recurrent optic neuritis[J]. Neurol Neuroimmunol Neuroinflamm, 2014, 1(4):e40.
[10]Cobo-Calvo A, Sepulveda M, Bernard-Valnet R, et al. Antibodies to myelin oligodendrocyte glycoprotein in aquaporin 4 antibody seronegative longitudinally extensive transverse myelitis: Clinical and prognostic implications[J]. Mult Scler, 2016, 22(3):312-319.
[11]Saadoun S, Waters P, Owens GP, et al. Neuromyelitis optica MOG-IgG causes reversible lesions in mouse brain[J]. Acta Neuropathol Commun, 2014, 2:35.
[12]Asavapanumas N, Ratelade J, Verkman AS, et al. Unique neuromyelitis optica pathology produced in naive rats by intracerebral administration of NMO-IgG[J]. Acta Neuropathol, 2014, 127(4):539-551.
[13]Jarius S, Ruprecht K, Kleiter I, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome[J]. J Neuroinflammation, 2016, 13(1):280.
[14]Yan Y, Li Y, Fu Y, et al. Autoantibody to MOG suggests two distinct clinical subtypes of NMOSD[J]. Sci China Life Sci, 2016, 59(12):1270-1281.
[15]Kim HJ, Paul F, Lana-Peixoto MA, et al. MRI characteristics of neuromyelitis optica spectrum disorder: an international update[J]. Neurology, 2015, 84(11):1165-1173.
[16]Jarius S, Kleiter I, Ruprecht K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 3: Brainstem involvement - frequency, presentation and outcome[J]. J Neuroinflammation, 2016, 13(1):281.
(本文编辑:邹晨双)
Clinical features of NMOSD patients with anti-MOG antibodies
LIXindi,WANGHuabing,ZHOUHeng,ZHOUanna,LIUYonghong,ZHAOlin,ZHANGXinghu*.
*DepartmentofNeurology,BeijingTiantanHospital,ChinaCapitalMedicalUniversity,Beijing100050,China
ZHANG Xinghu,Email: xhzhtiantan@hotmail.com
Objctive This study aimed to describe the clinical features of NMOSD patients with anti-MOG-antibodies to assist differential diagnoses in clinical practice. Methods Data including the patients’ clinical information and image findings from 32 patients with MS or NMOSD were collected and analyzed retrospectively. An enzyme linked immunosorbent assay was performed to detect the MOG antibodies in CSF. Cell based assays were used to detect AQP4 antibodies in the serum. Results Totally 29 NMOSD patients including 11 with AQP4 antibodies and 8 with MOG antibodies were enrolled. 4 (36.4%) AQP4 antibodies positive and 5 (62.5%) MOG antibodies positive NMOSD patients (excluding patients with both positive MOG-Abs and AQP4-Abs), as well as 1(7.7%)MS patient presented with myelitis and optic neuritis. The median Expanded Disability Status Scale (EDSS) score of NMOSD patients with MOG antibodies was 3.50 (2.50, 4.00), which was lower than the AQP4 antibodies positive group during the remission stage [4.00(3.50,6.00),Z=-2.379,P=0.020]. About 50% of MOG-Ab positive NMOSD patients had multiple lesions in the spinal cord, while all AQP4-Ab positive NMOSD patients had one lesion in the spinal cord (Z=6.967,P=0.018). The medium length of the spinal cord lesions in the AQP4-Ab positive group was 3(2,3), which was shorter than the AQP4-Ab positive group[4(3,5),Z=-2.499,P=0.012], and longer than the MS group [1.25(1,1.5),Z=-3.447,P<0.001]. 62.5% of the NMOSD patients with positive MOG-Ab had brain lesions, 3 of which were presented with NMOSD-like lesions, others were presented with MS-like lesions. Conclusions MOG-Ab positive NMOSD patients always combined with ON, lower EDSS and better prognosis. The spinal cord lesions in MOG-Ab positive NMOSD patients were multiple longitudinally extensive lesions. But the brain lesions lack specificity for differential diagnosis.
neuromyelitis optica spectrum disorder; multiple sclerosis; aquaporin 4; myelin oligodendrocyte glycoprotein; magnetic resonance imaging
10.3969/j.issn.1006-2963.2017.04.004
100050首都医科大学附属北京天坛医院 神经病学中心神经感染免疫科
张星虎,Email: xhzhtiantan@hotmail.com
R744.5+3
A
1006-2963(2017)04-0249-07
2017-02-06)
