改良的QuEChERS与HPLC-HESI/MS/MS同时测定中华鳖组织中氯硝柳胺和酰胺醇类药物及其代谢物的残留量
2017-08-31刘永涛杨秋红余琳雪杨移斌艾晓辉
刘永涛,董 靖,杨秋红,余琳雪,杨移斌,何 力,艾晓辉*
(1.中国水产科学研究院 长江水产研究所,湖北 武汉 430223;2.淡水水产健康养殖湖北省协同创新中心,湖北 武汉 430070;3.上海海洋大学 水产与生命学院,上海 201306)
改良的QuEChERS与HPLC-HESI/MS/MS同时测定中华鳖组织中氯硝柳胺和酰胺醇类药物及其代谢物的残留量
刘永涛1,2,董 靖1,2,杨秋红1,2,余琳雪3,杨移斌1,2,何 力1,艾晓辉1,2*
(1.中国水产科学研究院 长江水产研究所,湖北 武汉 430223;2.淡水水产健康养殖湖北省协同创新中心,湖北 武汉 430070;3.上海海洋大学 水产与生命学院,上海 201306)
建立了中华鳖(Trionyxsinensis)组织(血浆、肌肉、裙边、肝脏和肾脏)中氯硝柳胺、氯霉素、甲砜霉素、氟苯尼考和氟苯尼考胺同时测定的高效液相色谱-加热电喷雾电离源串联质谱法(HPLC-HESI/MS/MS)。样品经改进的QuEChERS方法提取净化,以氨化乙腈为提取剂,十八烷基硅烷键合硅胶(C18)粉为净化剂,甲醇-水为流动相,流速为0.3 mL/min,以Waters Symmetry®C18(2.1 mm×100 mm,3.5 μm)为色谱分离柱,采用正负离子分段扫描和多反应监测模式(MRM)检测。氯霉素、甲砜霉素、氟苯尼考和氟苯尼考胺采用内标标准曲线法定量,氯硝柳胺采用基质匹配标准曲线外标法定量。结果表明,在0.3~100 μg/L范围内,5种待测物均呈良好的线性关系,相关系数(r2)均不小于0.998 7。在1~20 μg/kg加标水平下,中华鳖空白血浆、肌肉、裙边、肝脏和肾脏的加标回收率为77.9%~105.3%(n=6),相对标准偏差为2.7%~10.5%(n=6),方法的检出限分别为0.5、0.1、0.5、0.5、0.5 μg/kg,定量下限分别为1.0、0.3、1.0、1.0、1.0 μg/kg。该方法操作简便、准确、灵敏度高,适用于中华鳖组织中氯硝柳胺、氯霉素、甲砜霉素、氟苯尼考和氟苯尼考胺残留量的同时测定。
中华鳖;氯硝柳胺;酰胺醇类;改良的QuEChERS;高效液相色谱-加热电喷雾电离源串联质谱
中华鳖(Trionyxsinensis),俗称甲鱼、团鱼,含高蛋白(高达43%)、低脂肪(仅为1.29%),维生素和矿物质的含量也较高,是深受人们喜爱的高档水产品[1]。近年来,随着消费需求量的增加,中华鳖的养殖规模不断扩大,养殖过程中发病也日趋增多,为了防治中华鳖的疾病,药物被大量使用,且滥用药物的情况时有发生。药物的不合理使用不仅会对环境造成污染,还会残留在动物源性食品中进而对人体健康产生危害。酰胺醇类药物包括氯霉素、甲砜霉素和氟苯尼考,氯霉素由于对人体的副作用如骨髓抑制和致癌性[2]而被中国、加拿大、美国、欧盟等国家和地区禁用[3],欧盟规定其在水产品中的最高残留限量(MRL)为0.3 μg/kg[4]。甲砜霉素、氟苯尼考在我国被批准用于水产动物疾病防治,氯硝柳胺在我国作为清塘剂用于杀灭养殖池塘内钉螺、椎实螺和野杂鱼[5]。欧盟规定甲砜霉素和氟苯尼考(以氟苯尼考胺为残留标示物)在有鳍鱼自然比例的皮肤和肌肉中的最高残留限量(MRL)分别为50 μg/kg和1 000 μg/kg[6],我国对鱼肌肉+皮中甲砜霉素和氟苯尼考的最高残留限量规定与欧盟相同[7]。目前,国内外尚未制定水产品中氯硝柳胺的残留限量标准,但由于其对鱼类高毒,且能引起DNA损伤[8],已引起人们的广泛关注。为保障中华鳖等水产品的食用安全,有必要建立一种简单、快速、准确和高灵敏度的同时测定中华鳖组织中氯硝柳胺和酰胺醇类抗生素及其代谢物残留量的方法。
目前尚未见水产品中氯硝柳胺和酰胺醇类抗生素及其代谢物同时测定的分析方法报道,更未见中华鳖组织中相关研究的报道。已有文献关于水产品中酰胺醇类抗生素测定的方法主要有高效液相色谱法[9]、气相色谱法[10-11]、气相色谱-质谱法[12]和液相色谱-质谱法[13-15],而水产品中氯硝柳胺残留量分析方法的报道相对较少[8,16-17],且大多采用传统的提取和净化技术,耗时且步骤复杂,另外还需大体积的提取溶剂。QuEChERS方法(快速、简单、便宜、有效、耐用和安全)[18]已被广泛用于食品安全领域,该方法样品制备过程简单、有效[3]。基于QuEChERS样品前处理方法,本文建立了中华鳖组织中氯硝柳胺、氯霉素、甲砜霉素、氟苯尼考和氟苯尼考胺同时测定的液相色谱-串联质谱法。
1 实验部分
1.1 仪器与试剂
TSQ Quantum Access Max液相色谱-质谱联用仪(Thermo Fisher Scientific公司);Hitachi 20PR-520型自动高速冷冻离心机(日本日立公司);Mettler-Toledo AE-240电子天平(梅特勒-托利多公司);FS-1高速匀浆机(华普达教学仪器有限公司);HQ-60-Ⅱ旋涡混合器(北京北方同正生物技术发展有限公司);HGC-12氮吹仪(HENGAO T&D公司)。
标准品:氯硝柳胺(纯度≥96%)、氯霉素(纯度≥98.6%)、甲砜霉素(纯度≥98.5%)、氟苯尼考(纯度≥99%)均购自德国Dr.Ehrenstorfer GmbH,氟苯尼考胺(纯度≥98%)、氘代氯霉素(d5-氯霉素)(纯度≥99%)、氘代甲砜霉素(d3-甲砜霉素,纯度≥98%)、氘代氟苯尼考(d3-氟苯尼考,纯度≥98%)均购自加拿大TRC公司;十八烷基硅烷键合硅胶(C18,40~60 μm,天津博纳艾杰尔科技有限公司),乙二胺-N-丙基硅烷(PSA,40~60 μm,天津博纳艾杰尔科技有限公司),多壁碳纳米管(MWCNTs)(纯度>95%,外径8~15 nm,长度<50 μm,北京博宇高科新材料技术有限公司);乙腈、甲醇、质谱水(色谱纯,美国J.T.Baker公司),氨水、无水硫酸钠、氯化钠(分析纯,上海国药集团试剂有限公司);氨化乙腈提取液的配制:1 000 mL乙腈中添加2 mL氨水,混匀即配制成0.2%的氨水乙腈提取液。
1.2 标准储备液及混合标准中间液的配制
分别准确称取适量氯硝柳胺、氯霉素、甲砜霉素、氟苯尼考、氟苯尼考胺标准品,用甲醇分别溶解并定容,配制成100 mg/L的标准储备液,于-20 ℃冰箱中保存。分别移取1 mL标准储备液于100 mL容量瓶中,用30%(体积分数)乙腈水溶液稀释成1 mg/L的混合标准中间液。分别准确称取适量d5-氯霉素、d3-甲砜霉素和d3-氟苯尼考标准品,用甲醇分别溶解并定容成10 mg/L内标储备液;分别移取1 mL内标储备液至100 mL容量瓶中用30%乙腈水溶液(体积比)稀释至100 mL,配制成100 μg/L混合内标中间液。
1.3 标准工作曲线及氯硝柳胺空白基质匹配标准曲线的绘制
用30%乙腈水溶液稀释混合标准中间液,得到含氯硝柳胺、氯霉素、甲砜霉素、氟苯尼考、氟苯尼考胺的质量浓度分别为0.3、0.5、1.0、5.0、10.0、20.0、50.0、100.0 μg/L,且含d5-氯霉素、d3-甲砜霉素和d3-氟苯尼考质量浓度均为2 μg/L的标准工作溶液,上HPLC-HESI/MS/MS分析,绘制标准工作曲线,计算回归方程与相关系数。
采用“1.4”方法制备中华鳖血浆、肌肉、裙边、肝脏和肾脏空白组织样品溶液,用空白组织样品溶液稀释混合标准中间液,制备氯硝柳胺质量浓度分别为0.5、1.0、5.0、10.0、20.0、50.0、100.0 μg/L的空白基质匹配标准溶液,上HPLC-HESI/MS/MS分析,绘制空白基质匹配标准工作曲线,计算回归方程与相关系数。
1.4 样品前处理
1.4.1 中华鳖血浆样品 移取1 mL血浆样品于10 mL离心管中,加入40 μL 100 μg/L的内标混合溶液,再加入4 mL氨化乙腈提取液,涡旋30 s后,加入2 g无水硫酸钠和0.5 g氯化钠,涡旋振荡1 min,7 000 r/min离心5 min,转移提取液至新的15 mL带刻度离心管中,残渣重复提取1次,合并提取液至同一15 mL离心管中,并用乙腈定容至10 mL,再加入300 mg C18粉,涡旋振荡30 s,7 000 r/min离心5 min,准确移取5 mL提取液至另一10 mL离心管中,于50 ℃氮吹至干,用1 mL 30%乙腈水溶液溶解残渣,涡旋振荡30 s,10 000 r/min,离心5 min,取离心后的液体过0.22 μm滤头,上HPLC-HESI/MS/MS分析。
1.4.2 中华鳖肌肉与裙边样品 称取2 g 肌肉或裙边样品于15 mL离心管中,加入40 μL 100 μg/L的内标混合溶液,加入7 mL氨化乙腈提取液,涡旋振荡30 s,再加入4 g 无水硫酸钠和1 g氯化钠,涡旋振荡1 min,7 000 r/min离心5 min,转移提取液至另一15 mL离心管中,重复提取1次,合并提取液,并用乙腈定容至15 mL,再加入300 mg C18粉,涡旋振荡30 s,7 000 r/min离心5 min,准确移取7.5 mL提取液至10 mL离心管中,于50 ℃氮吹至干,用1 mL 30%乙腈水溶液溶解残渣,涡旋振荡30 s,10 000 r/min离心5 min,取离心后的液体过0.22 μm滤头,上HPLC-HESI/MS/MS分析。
1.4.3 中华鳖肝脏与肾脏样品 称取1 g 肝脏或肾脏样品,置于10 mL离心管中,加入40 μL 100 μg/L的内标混合溶液,再加入5 mL氨化乙腈提取液,涡旋振荡30 s,再加入2 g无水硫酸钠和0.5 g氯化钠,涡旋振荡1 min,7 000 r/min离心5 min,将提取液转移至另一10 mL离心管中,重复提取1次,合并提取液,并用乙腈定容至10 mL,再加入300 mg C18粉,涡旋振荡30 s,7 000 r/min离心5 min,准确移取5 mL提取液于10 mL离心管中,于50 ℃氮吹至干,用1 mL 30%乙腈水溶液溶解残渣,涡旋振荡30 s,10 000 r/min离心5 min,取离心后的液体过0.22 μm滤头,上HPLC-HESI/MS/MS分析。
1.5 色谱-质谱分析条件
色谱柱:Waters Symmetry®C18(2.1 mm×100 mm,3.5 μm);柱温:35 ℃,流速:0.3 mL/min;流动相:A为甲醇,B为水;梯度洗脱条件:初始条件20% A;0~3.0 min,20%~90% A;3.0~5.8 min,90% A;5.8~5.9 min,90%~20% A;5.9~7.0 min,20% A;进样量10 μL。
离子化模式:加热大气压电喷雾离子源(HESI),正离子、负离子分段(Segment)多反应监测模式(MRM)进行检测,Segment 1:0~1.2 min,正离子扫描,Segment 2:1.2~5.0 min,负离子扫描;Segment 3:5.0~7.0 min,负离子扫描。正离子模式喷雾电压:3 500 V,负离子模式喷雾电压:-3 000 V,源内解离电压 0 V,蒸发气温度:300 ℃,鞘气压力40 arb,辅助气压力10 arb,碰撞气及压力:氩气,1.5 mTorr,离子传输毛细管温度:350 ℃,Q1 PW 0.4,Q3 PW 0.7。
2 结果与讨论
2.1 色谱-质谱条件的优化
2.1.1 质谱条件的优化 分别将10 mg/L的氯硝柳胺、氯霉素、甲砜霉素、氟苯尼考、d5-氯霉素、d3-甲砜霉素、d3-氟苯尼考和氟苯尼考胺标准溶液通过注射器注入质谱仪,对其母离子进行扫描,发现氯硝柳胺、氯霉素、甲砜霉素、氟苯尼考、d5-氯霉素、d3-甲砜霉素和d3-氟苯尼考在负离子模式下产生更多的[M-H]-分子离子,这与该类药物均含有高电负性的卤素原子和羟基基团有关[18],母离子的m/z分别为324.9、320.9、354.0、355.9、325.9、357.0和358.7,而氟苯尼考胺则在正离子模式下产生大量的[M+H]+分子离子,其母离子的m/z为248.0,因此,氯硝柳胺、氯霉素、甲砜霉素、氟苯尼考、d5-氯霉素、d3-甲砜霉素和d3-氟苯尼考适合在负离子模式下进行分析,而氟苯尼考胺则适合在正离子模式下进行分析。在待测物适宜的离子模式下,以子离子扫描方式进行二级质谱分析,分别找出其定量和定性子离子以及对应的碰撞能量,结果见表1。HESI与ESI相比,采用设定蒸发气的温度来提高辅助气的温度,起到了加热的作用,因而离子化效率和重现性比ESI源好,且灵敏度有所提高[19],本研究发现设定HESI源蒸发气的温度为300 ℃时,较不设蒸发气温度采用ESI的方式分析待测物,其灵敏度可提高约2倍。

表1 氯硝柳胺、氯霉素、甲砜霉素、氟苯尼考、氟苯尼考胺、d5-氯霉素、d3-甲砜霉素和d3-氟苯尼考的质谱条件
*quantitation ion

图1 5种待测物及内标混合标准溶液的多反应监测色谱图(0.5 μg/L)Fig.1 MRM chromatograms of five analytes and internal standard mixed standard solution(0.5 μg/L)1.FFA(m/z 248.0>230.0),2.NIC(m/z 324.9>289.0),3.CAP(m/z 320.9>152.0),4.d5-CAP(m/z 325.9>157.0),5.TAP(m/z 354.0>184.9),6.d3-TAP(m/z 357.0>188.0),7.FF(m/z 355.9>335.9),8.d3-FF (m/z 358.7>338.8)
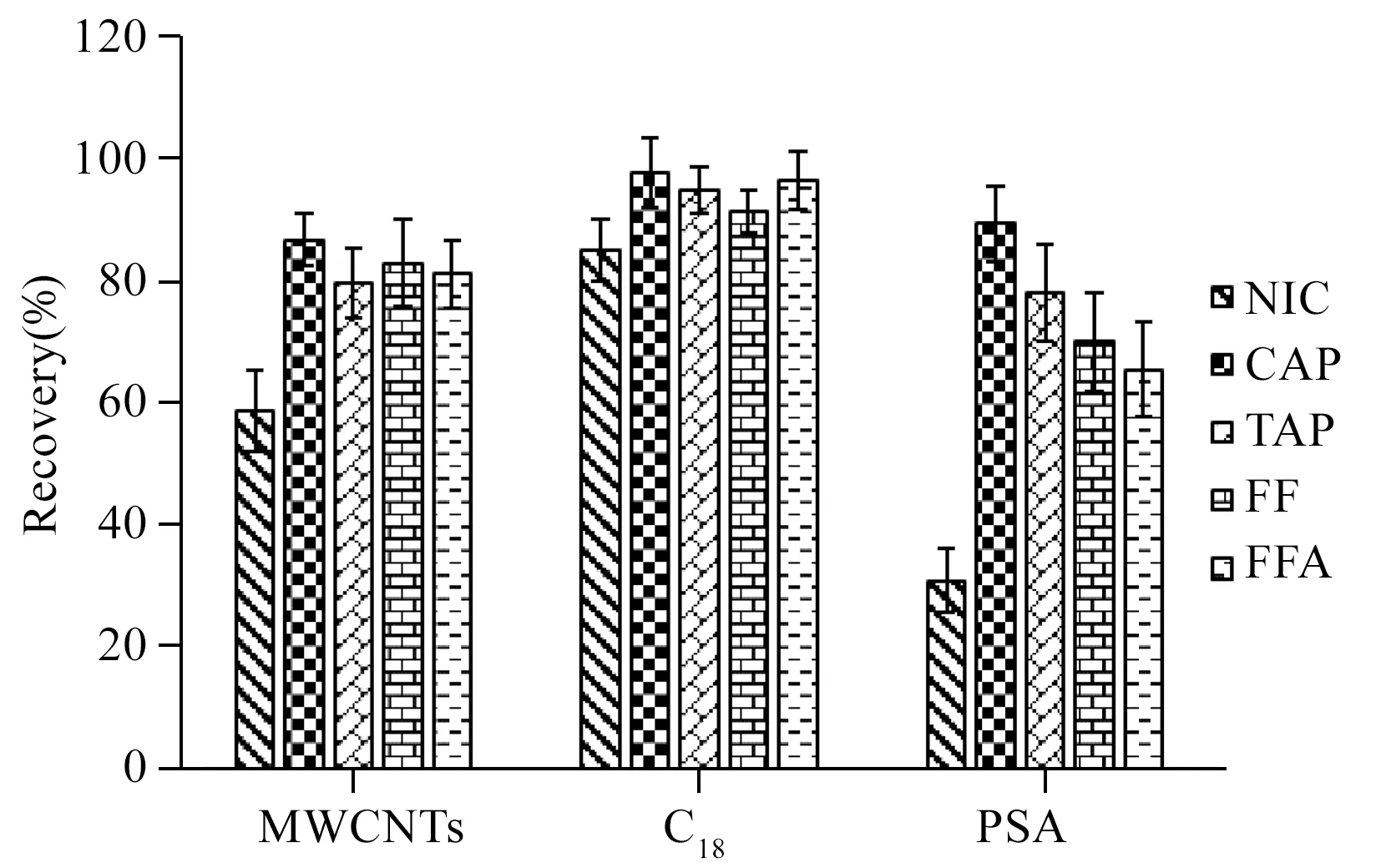
图2 不同吸附剂对氨化乙腈提取液的净化作用Fig.2 Effects of different sorbents in ammoniated acetonitrile extraction solution
2.1.2 色谱条件的优化 流动相中加入甲酸或乙酸均可提高氟苯尼考胺的质谱响应度,但会抑制氯硝柳胺、氯霉素、甲砜霉素和氟苯尼考的质谱响应度,而氯霉素属禁用药物,对其灵敏度的要求较氟苯尼考胺更高。此外,氯硝柳胺的pKa值为7.3,为两性化合物[16],当流动相中加入甲酸、乙酸或氨水时氯硝柳胺的保留时间易发生漂移,且影响其离子化效率的稳定性,因此本实验最终选择甲醇-水作为流动相,以获得最佳的分离效果。5种待测化合物的极性由大到小顺序为氟苯尼考胺>甲砜霉素>氟苯尼考>氯霉素>氯硝柳胺,国家标准GB/T 22959-2008[20]采用40%甲醇和水为流动相,可在Supelco Discovery C18(150 mm×2.1 mm,5 μm)色谱柱上对氯霉素、甲砜霉素和氟苯尼考进行分离,而本研究采用该流动相时很难将氯硝柳胺从反相色谱柱上洗脱,因此最终选择采用“1.5”所述梯度洗脱程序分离洗脱5种待测物(见图1)。
2.2 样品前处理条件的优化
2.2.1 提取溶液的选择 动物组织中氯霉素、甲砜霉素和氟苯尼考的常用提取剂有乙酸乙酯、乙腈、丙酮和二氯甲烷等溶剂,但上述溶剂并不能同时提取氯硝柳胺、氟苯尼考胺、氯霉素、甲砜霉素和氟苯尼考,因氟苯尼考胺是碱性化合物,更易溶解在高pH值的溶液中[19],而碱性提取液也有利于氯硝柳胺的提取[8],由于乙腈的脂质提取率低,且基质离子化作用小[21],因此本实验最终选用氨化乙腈作为提取溶剂,并参考Liu等[8]对氨化乙腈溶液的制备方法进行配制。结果表明,以氨化乙腈作为提取剂可使水产品中的氯硝柳胺、氯霉素、甲砜霉素、氟苯尼考和氟苯尼考胺残留获得较好的提取效率。
2.2.2 净化用吸附剂的选择及其用量的确定 在中华鳖空白肌肉组织中添加混合标准溶液,使其加标浓度为5 μg/kg。按照“1.4.2”方法进行提取后,分别考察了200 mg的多壁碳纳米管(MWCNTs)、乙二胺-N-丙基硅烷(PSA)和十八烷基硅烷键合硅胶(C18)作为净化剂时的回收率。由图2可知,C18对样品提取液的净化效果最优,PSA的净化效果最差。这是由于PSA吸附剂是在硅胶上键合了乙二胺-N-丙基官能团,是与—NH2相似的吸附剂,对5种待测物均有一定的保留作用且对氯硝柳胺的保留作用最大,MWCNTs是一种具有较强吸附作用的纳米材料,性质与C18相似[21-23],对5种待测物也有一定的吸附,因此选择C18作为净化剂。进一步对C18的用量进行考察,结果显示不同用量C18对氨化乙腈提取液的净化效果为300 mg>200 mg>100 mg,因此,实验选择C18的用量为300 mg。
2.3 基质效应
液相色谱-质谱联用技术在生物样品分析中普遍存在基质效应。氘代同位素内标与待测物结构相似,在色谱和质谱上的特征相似,不但可抵消质谱离子化时的基质效应,还可消除样品前处理过程中的差异[24]。本研究采用d5-氯霉素、d3-甲砜霉素和d3-氟苯尼考作为内标,对中华鳖组织中氯霉素、甲砜霉素、氟苯尼考和氟苯尼考胺进行内标法定量,可消除基质效应对定量结果准确性的影响。由于未购买到氯硝柳胺的同位素标准品,采用空白基质提取液配制与标准溶液相同浓度的基质匹配标准溶液(1、5、20 μg/L),分别测定后按照Liu等[8]方法计算中华鳖组织对氯硝柳胺产生的基质效应,结果表明,中华鳖组织对氯硝柳胺均产生较强的基质抑制作用,其中中华鳖血浆对氯硝柳胺的基质效应分别为-11.2%、-9.8%和-9.5%,中华鳖肌肉组织对氯硝柳胺的基质效应分别为-12.6%、-13.7%和-10.4%,中华鳖裙边对氯硝柳胺的基质效应分别为-11.5%、-12.7%和-13.2%,中华鳖肝脏组织对氯硝柳胺的基质效应分别为-13.3%、-14.9%和-12.4%,中华鳖肾脏组织对氯硝柳胺的基质效应分别为-14.3%、-12.9%和-11.9%。因此,本文采用空白基质匹配校正曲线的方法对氯硝柳胺进行定量以减少测定误差。
2.4 线性关系
2.4.1 标准工作曲线 将“1.3”制备的标准工作溶液进样分析,以氯硝柳胺的质量浓度(x,μg/L)为横坐标,峰面积(A)为纵坐标,绘制标准工作曲线;分别以氯霉素、甲砜霉素、氟苯尼考和氟苯尼考胺的质量浓度(x,μg/L)为横坐标,对应峰面积分别与d5-氯霉素、d3-甲砜霉素、d3-氟苯尼考和d3-氟苯尼考峰面积的比值(y)为纵坐标,分别绘制标准工作曲线。结果表明,氯硝柳胺、氯霉素、甲砜霉素、氟苯尼考和氟苯尼考胺均在0.3~100 μg/L浓度范围内呈线性相关,回归方程分别为A=48 793x+60 791,y=3.569 7x-1.583 1,y=0.420 3x-0.074 8,y=0.496 2x+0.336 6和y=0.636 9x+0.401 8,相关系数分别为0.998 7、0.999 2、0.999 8、0.999 6和0.999 2。
2.4.2 氯硝柳胺的空白基质匹配标准曲线 将“1.3”制备的中华鳖血浆、肌肉、裙边、肝脏和肾脏空白基质匹配标准溶液进样分析,以氯硝柳胺的质量浓度(Cm)为横坐标,峰面积(Ai)为纵坐标,绘制空白基质匹配标准工作曲线。结果表明,氯硝柳胺在0.5~100 μg/L浓度范围内呈线性相关,基质匹配标准曲线、相关系数和线性范围见表2。

表2 氯硝柳胺的基质匹配标准曲线、相关系数和线性范围
2.5 方法的回收率、精密度、检出限与定量下限
在中华鳖空白血浆、肌肉、裙边、肝脏和肾脏中添加浓度水平为1.0、5.0、20 μg/kg的氯硝柳胺、氯霉素、甲砜霉素、氟苯尼考和氟苯尼考胺混合标准溶液,每个浓度水平平行6个样品,按照“1.5”方法进行样品前处理,在“1.6”色谱条件下进行测定,得3个加标水平下中华鳖组织中的平均回收率为77.9%~105.3%,相对标准偏差(RSD)为2.7%~10.5%。氯硝柳胺、氯霉素、甲砜霉素、氟苯尼考和氟苯尼考胺在中华鳖组织中以大于3倍信噪比计算的检出限(LOD)分别为0.5、0.1、0.5、0.5、0.5 μg/kg,以大于10倍信噪比计算的定量下限(LOQ)分别为1.0、0.3、1.0、1.0、1.0 μg/kg。说明本方法可以满足中华鳖组织中氯硝柳胺、氯霉素、甲砜霉素、氟苯尼考和氟苯尼考胺的检测要求。方法回收率、精密度、检出限和定量下限结果见表3。
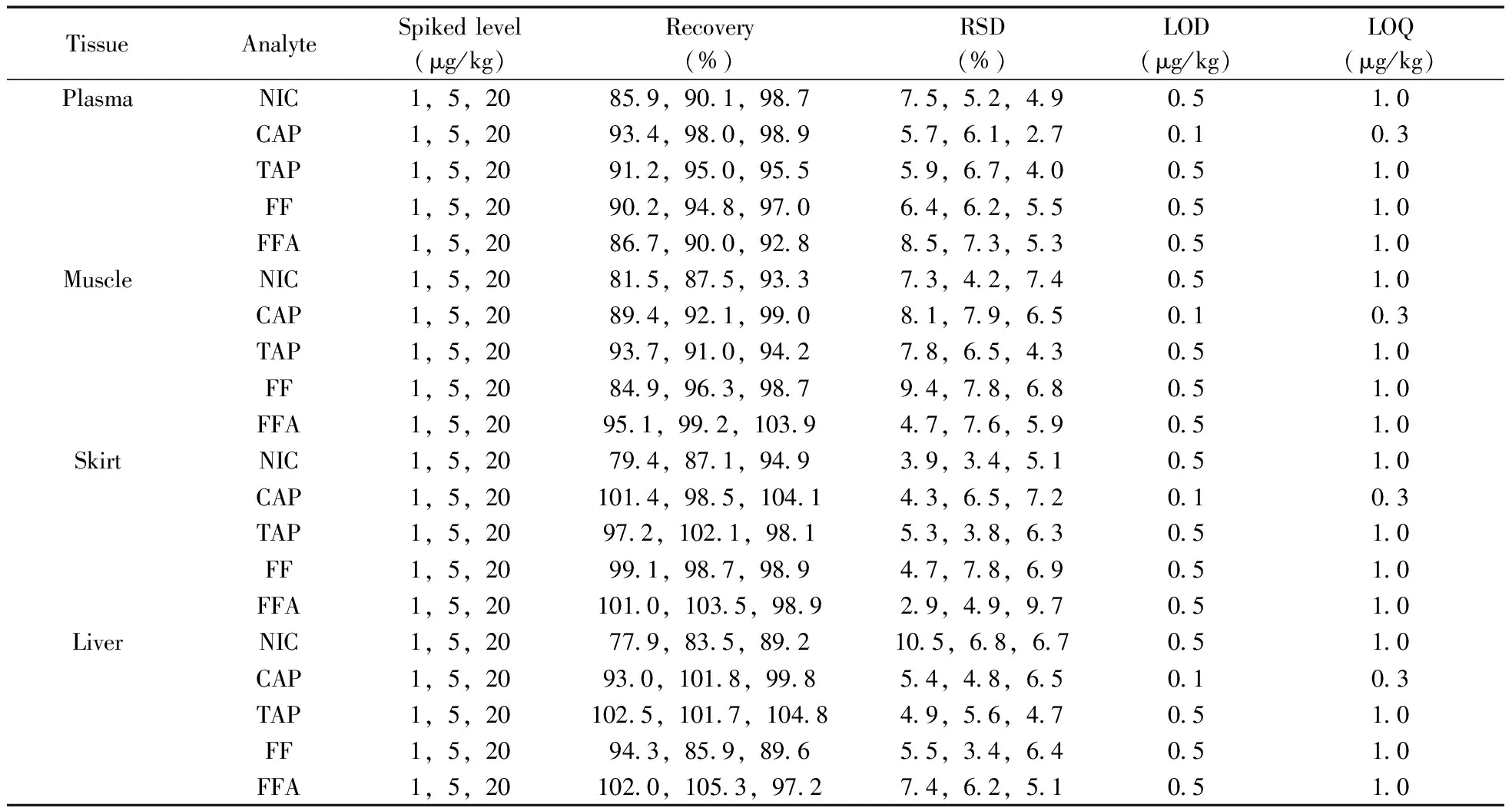
表3 中华鳖组织中5种目标物的加标回收率、相对标准偏差、检出限和定量下限
(续表3)

TissueAnalyteSpikedlevel(μg/kg)Recovery(%)RSD(%)LOD(μg/kg)LOQ(μg/kg)KidneyNIC1,5,2081 9,87 9,97 26 8,5 7,6 10 51 0CAP1,5,20100 5,104 8,105 15 6,6 0,7 10 10 3TAP1,5,2098 5,103 0,99 45 3,6 5,5 10 51 0FF1,5,20103 2,100 9,97 910 1,6 9,7 50 51 0FFA1,5,20101 9,95 2,98 65 1,6 7,4 60 51 0
2.6 方法的应用
将该方法用于市售5份中华鳖、3份黄颡鱼、4份团头鲂肌肉样品,以及送检的怀疑氯硝柳胺中毒死亡的白鲢和草鱼肌肉样品的测定,结果表明市售的1份中华鳖肌肉中检出甲砜霉素,其残留量仅为2.41 μg/kg,远低于我国现行规定的鱼中甲砜霉素的最大残留限量值(50 μg/kg),而送检的怀疑中毒的白鲢和草鱼肌肉样品中分别检出高含量的氯硝柳胺,其含量分别为94.51 μg/kg和68.75 μg/kg。
3 结 论
本文采用改良的QuEChERS前处理方法,建立了中华鳖组织(血浆、肌肉、裙边、肝脏和肾脏)中氯硝柳胺、氯霉素、甲砜霉素、氟苯尼考和氟苯考胺残留的正负离子切换HPLC-HESI/MS/MS同时测定的分析方法。该方法样品前处理简单快速,灵敏度高,回收率和精密度良好,方法的定量下限均能满足各国对最大残留限量值的要求。从而为开展氯硝柳胺和酰胺醇类药物在中华鳖体内代谢研究和残留量监测提供了技术支撑,也可为水产养殖中由氯硝柳胺引起的鱼塘中毒事件提供一种有效的分析判断方法。
[1] Tan A P,Zhao F,Jiang L,Luo L,Wang W L,Peng H L,Chen Y L,Zou W M.GuangdongAgric.Sci.(谭爱萍,赵飞,姜兰,罗理,王伟利,彭华林,陈永乐,邹为民.广东农业科学),2011,(20):115-119.
[2] Yuan M,Sheng W,Zhang Y,Wang J P,Yang Y J,Zhang S G,Goryacheva I Y,Wang S.Anal.Chim.Acta,2012,751(21):128-134.
[3] Liu H Y,Lin S L,Fuh M R.Talanta,2016,150:233-239.
[4] European Commission Decision 2003/181/EC of March 2003 on Amending Decision 2002/657/EC as Regard the Setting of Minimum Required Performance Limits(MRPLs) for Certain Residues in Food Animal Origin.Off.J.Eur.Commun.,2003,L71:17.
[5] China Institute of Veterinary Drugs Control.National Standard Collection of Veterinary Medicine-Local Standards for Veterinary Medicine Increased National Standards(The First Volume).Beijing:China Agriculture Press(农业部兽药评审中心.兽药国家标准汇编-兽药地方标准上升国家标准(第一册).北京:中国农业出版社),2010:70,121,145.
[6] European Commission Regulation(EU) No 37/2010 of 22 December 2009 on Pharmacologically Active Substances and Their Classification Regarding Maximum Residue Limits in Foodstuffs of Animal Origin.Off.J.Eur.Commun.,2010,L15:32,64.
[7] Ministry of Agriculture.No.235 Bulletin of the Ministry of Agriculture of the People’s Republic of China(农业部.中华人民共和国农业部公告第235号).[2008-06-29].http://yz.hz-agri.gov.cn/uploadFiles/2005-10/1130221564406.doc.
[8] Liu Y T,Ai X H,Wang F H,Suo W W,Yang Q H,Yang H,Xu N.Anal.Lett.,2015,48(3):929-943.
[9] Yang F,Chen G N.FujianAnalysis&Testing(杨方,陈国南.福建分析测试),2005,14(1):2112-2113.
[10] Liu Y T,Li R,Yuan K P,Yang H,Ai X H.FreshwaterFisheries(刘永涛,李荣,袁科平,杨红,艾晓辉.淡水渔业),2007,37(2):44-47.
[11] Yang Q H,Ai X H,Li R,Liu Y T.Chin.J.Anal.Lab.(杨秋红,艾晓辉,李荣,刘永涛.分析试验室),2015,34(5):533-537.
[12] Shao H,Leng K L,Zhou M Y,Gao H,Sun W H,Xing L H,Miao J K,Liu K.ProgressinFisherySciences(邵会,冷凯良,周明莹,高华,孙伟红,邢丽红,苗钧魁,刘坤.渔业科学进展),2015,36(3):137-141.
[13] Tao X C,Huang H,Liao J M,Gao P,Huang G F,Zeng D D.J.Chin.Inst.FoodSci.Technol.(陶昕晨,黄和,廖建萌,高平,黄国芳,曾丹丹.中国食品学报),2014,14(1):232-238.
[14] Peng L,Guo D W,Zhang W,Ruan X C,Gao X G,Gao Y,Li J J,Zheng Y N,Ji H,Jiang S X.AnimalHusbandryandVeterinaryMedicine(彭麟,郭大伟,张伟,阮祥春,高修歌,高颖,李娇娇,郑亚妮,季辉,江善祥.畜牧与兽医),2016,48(4):30-35.
[15] Pan X D,Wu P G,Jiang W,Ma B J.FoodControl,2015,52:34-38.
[16] Schreier T M,Dawson V K,Cho Y,Spanjers N J,Boogaard M A.J.Agric.FoodChem.,2000,48:2212-2215.
[17] Caldow M,Sharman M,Kelly M,Day J,Hird S,Tarbin J A.J.Chromatogr.A,2009,1216(46):8200-8205.
[18] Anastassiades M,Lehotay S J.J.AOACInt.,2003,86(2):412-431.
[19] Pan H L,Lin L S,Ding Y F,Chen X Y,Zhong D F.ActaPharm.Sin.(潘华玲,林丽珊,丁珏芳,陈笑艳,钟大放.药学学报),2014,49(1):95-100.
[20] Xie K Z,Jia L F,Yao Y L,Xu D,Chen S Q,Xie X,Pei Y,Bao W B,Dai G J,Wang J Y,Liu Z P.J.Chromatogr.B,2011,879:2351-2354.
[21] Zhao H X,Sun Y H,Ding M Y,Chen L M,Deng W,Zhao M B.J.Instrum.Anal.(赵海香,孙艳红,丁明玉,陈丽梅,邓维,赵孟彬.分析测试学报),2011,30(6):635-639.
[22] Zhao H X,Liu H P,Yan Z Y.Chin.J.Chromatogr.(赵海香,刘海萍,闫早婴.色谱),2014,32(3):294-298.[23] Zhao M Y,Han F,Sun J W,Song W,Lü Y N,Hu Y Y,Zheng P,Sheng X,Deng X J.J.Instrum.Anal.(赵暮雨,韩芳,孙锦文,宋伟,吕亚宁,胡艳云,郑平,盛旋,邓晓军.分析测试学报),2016,35(12):1513-1520.[24] Xiang P,Shen M,Zhuo X Y.J.Instrum.Anal.(向平,沈敏,卓先义.分析测试学报),2009,28(6):753-756.
武汉病毒所在抗病毒免疫研究方面获得重要突破
中国科学院武汉病毒研究所周溪研究员课题组与军事医学科学院微生物流行病研究所秦成峰研究员课题组合作,在抗病毒免疫研究方面取得重要进展,揭示了RNA干扰(RNAi)通路在哺乳动物中具有抗病毒免疫功能。相关研究成果以“Human virus-derived small RNAs can confer antiviral immunity in mammals”(人类病毒来源的小RNA在哺乳动物中产生抗病毒免疫反应)为题发表在Immunity上。
RNAi是一种在真核生物中高度保守的转录后基因沉默机制, 并已被公认在真菌、植物和无脊椎动物中起到关键的抗病毒免疫作用。在RNAi抗病毒过程中, 病毒RNA复制所产生的双链RNA(dsRNA)被宿主Dicer蛋白识别并切割成小干扰RNA(siRNA)。这些病毒衍生的siRNAs(vsiRNAs)被转移到 RNA 诱导沉默复合体 (RISC), 并介导同源病毒RNA的降解,从而达到抗病毒的目的。尽管RNAi在哺乳动物中也保守存在,并被广泛用于生命科学与技术研究,然而,在哺乳动物中,RNAi是否同样能起到抗病毒免疫作用仍不清楚。
在该研究中,研究者利用人肠道病毒71型(EV71)感染的人类体细胞及小鼠为模型,发现其非结构蛋白3A具有RNAi抑制子(VSR)功能。3A能够通过与病毒dsRNA结合来阻止Dicer对其剪切,抑制vsiRNAs的产生。当3A的VSR活性被缺失,VSR缺陷型EV71病毒能在细胞与小鼠中激发RNAi反应,并产生大量vsiRNAs。这些vsiRNA通过Dicer剪切病毒dsRNA产生、被装配进RISC、并高效地介导同源病毒RNA的降解。在正常的人体细胞和小鼠中,VSR缺陷型病毒的复制被极大地抑制;而在RNAi通路缺失的细胞中,突变病毒的复制得到显著的拯救。同时,研究者们还证明RNAi在哺乳动物中所发挥的抗病毒作用不依赖于干扰素反应。
该研究在人类体细胞及动物水平发现了病毒感染可以产生具抗病毒功能的vsiRNA,确证了RNAi在哺乳动物中是一条抗病毒天然免疫通路;同时,也揭示了一种人类病毒在逃逸RNAi天然免疫的具体机制。该工作完善了对哺乳动物抗病毒免疫机制的认识,并为该领域的后续研究以及针对该通路的抗病毒药物设计或免疫疗法研究提供了理论基础。
(信息来源:中国科学院武汉病毒研究所)
Determination of Niclosamide and Amphenicols Residues in Chinese Soft-shelled Turtle(Trionyxsinensis) Tissues by Modified QuEChERS Combined with HPLC-HESI/MS/MS
LIU Yong-tao1,2,DONG Jing1,2,YANG Qiu-hong1,2,YU Lin-xue3,YANG Yi-bin1,2,HE Li1,AI Xiao-hui1,2*
(1.Yangtze River Fisheries Research Institute,Chinese Academy of Fishery Sciences,Wuhan 430223,China;2.Hubei Freshwater Aquaculture Collaborative Innovation Center,Wuhan 430070,China;3.College of Fisheries and Life Science,Shanghai Ocean University,Shanghai 201306,China)
A modified QuEChERS sample preparation combined with high performance liquid chromatography-heated electrospray ionization-tandem mass spectrometric(HPLC-HESI/MS/MS) method was established for the simultaneous determination of niclosamide(NIC),chloramphenicol(CAP),thiamphenicol(TAP),florfenicol(FF) and florfenicol amine(FFA) in plasma,muscle,skirt,liver and kidney of Chinese soft-shelled turtle(Trionyxsinensis).The sample was extracted and purified using a modified QuEChERS method,and ammoniate acetonitrile and octadecyl silane(C18) were chosen as extractant and purification,respectively.The mobile phase comprised of methanol and water,and the flow rate was set at 0.3 mL/min.The sample prepared was separated on a Waters Symmetry®C18(2.1 mm×100 mm,3.5 μm) column,and determined in the positive and negative ion multiple reaction monitoring mode through polarity switching between time segments.CAP,TAP,FF and FFA were quantified by internal standard calibration curve method,and NIC was used the external standard matrix-matched standard curve method.Good linearities were observed in the range of 0.3-100 μg/L for five analytes,with correlation coefficients not more than 0.998 7.The average recoveries of 5 analytes at the spiked level of 1-20 μg/kg in plasma,muscle,skirt,liver and kidney of Chinese soft-shelled turtle(Trionyxsinensis) ranged from 77.9% to 105.3% with relative standard deviations(RSD) of 2.7%-10.5%.The detection limits(LOD) for NIC,CAP,TAP,FF and FFA in Chinese soft-shelled turtle tissues were 0.5,0.1,0.5,0.5,0.5 μg/kg,and the quantitation limits(LOQ) were 1.0,0.3,1.0,1.0,1.0 μg/kg,respectively.The method is simple,accurate,highly sensitive,and is suitable for the simultaneous determination of NIC,CAP,TAP,FF and FFA residues in Chinese soft-shelled turtle tissues.
Chinese soft-shelled turtle;niclosamide;amphenicols;modified QuEChERS;high performance liquid chromatography-heated electrospray ionization-tandem mass spectrometry(HPLC-HESI/MS/MS)
2017-04-05;
2017-04-15
中国水产科学研究院基本科研业务费资助项目(2016JBF0104);现代农业人才支撑计划项目(2016-2020)
10.3969/j.issn.1004-4957.2017.08.002
O657.63;R978.1
A
1004-4957(2017)08-0955-08
*通讯作者:艾晓辉,博士,研究员,研究方向:水产动物药理,Tel:027-81780298,E-mail:thincat2005@sina.com
