多巴反应性肌张力障碍2例报告并文献复习
2017-08-17江转南孟哲侯乐乐刘祖霖欧辉张丽娜梁立阳
江转南孟哲侯乐乐刘祖霖欧辉张丽娜梁立阳
·病例报告·
多巴反应性肌张力障碍2例报告并文献复习
江转南*孟哲*侯乐乐*刘祖霖*欧辉*张丽娜*梁立阳*
多巴反应性肌张力障碍 左旋多巴 酪氨酸羟化酶缺乏症
多巴反应性肌张力障碍 (dopa-responsive dystonia,DRD)是一种非常罕见的遗传性进行性肌张力障碍疾病,其临床症状十分复杂,导致临床早期治疗极困难,本文报道2例散发病例并结合文献回顾分析,加深临床医生对该病的发病机制、临床表现及遗传学基础的了解。
1 临床资料
例1,患者,女,2岁5个月。以“四肢震颤、运动倒退1年半”为主诉于2015年7月入住我院。患儿7月龄出现四肢震颤,以双下肢为甚,双手握物时震颤明显,动作缓慢,症状呈晨轻暮重。8月龄于呼吸道感染后出现运动倒退,表现为不能翻身,抬头、独坐不稳,四肢震颤加重。体格检查:12 kg(P25),头围:48.5 cm(P25),身长:89 cm(P25),神志清楚,心肺腹部查体无特殊(P25、P50、P75:儿童生长发育曲线指标)。动作反应慢,四肢肌张力高,以上肢明显,时呈齿轮样增高,时呈铅管样增高,双侧巴氏征(+-),双侧踝阵挛(+),双踝外翻。曾于外院行视觉诱发电位提示右侧轻度异常,头颅MR、脑电图、肌电图、血尿遗传代谢病筛查、脊肌萎缩症SMN1基因测序均未见明显异常。外院诊断为“脑性瘫痪”,予规律理疗、针灸等治疗,症状无改善。G1P1,出生史、家族史无特殊。生后3个月抬头稳,4~5个月翻身可,6个月双手撑地可独坐。入院后查头颅MR、血常规、肝肾功能、电解质未见异常。全外显子基因测序提示患儿存在TH基因杂合致病突变,随后对其家系进行TH基因检测发现其父母均为致病突变携带者(如图1),确诊为DRD后予美多芭治疗每次0.0625 g,每日3次,患儿出现明显异动症,美多芭调整剂量至每次0.03 g,每日3次,辅以苯海索片(每次2 mg,每日3次)、氯丙嗪(每次8 mg,每晚1次)调节运动,患儿异动症缓解。1个月后患儿运动、语言能力逐渐改善,6个月后独走稳,四肢震颤消失,能画画,能与人简单交流,会唱歌、背诗。
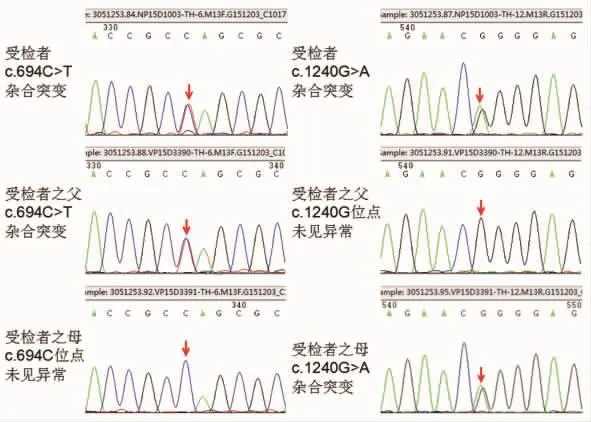
图1 例1患儿及其父母的基因检测结果
例2,患者,女,2岁3个月。以“发现运动倒退1年”为主诉于2016年9月入住我院。患儿生后5月龄可抬头,8月龄抓物稳,可逗笑、追物,8月、12月龄于呼吸道感染后出现运动倒退,表现为不能抬头、四肢软瘫、握物不稳,症状呈晨轻暮重。体格检查:11.6 kg(P25),头围:48 cm(P50),身长:89.5 cm(P50),神志清楚,心肺腹部查体无特殊。动作反应慢,四肢肌张力偏低,以近端明显,双侧巴氏征(-),双侧膝反射(++),双侧踝阵挛未引出。曾于外院行头颅MR示双侧侧脑室额角稍扩张。外院诊断 “脑性瘫痪”,予针灸、理疗等治疗,症状无明显改善。G2P2,31+周,生后于固尔苏、呼吸机辅助呼吸,家族史无特殊。入院检查结果:脊肌萎缩症SMN1基因测序、血常规、粪便常规、尿常规、IGF-1、IGFBP-3、单纯疱疹病毒IgM抗体、弓形虫IgM抗体、风疹病毒IgM抗体、巨细胞病毒IgM抗体、肝肾功能、电解质未见明显异常。全外显子基因测序提示TH基因杂合突变,随后对其家系进行TH基因检测发现其父母均为TH基因突变携带者(如图2),其中其父携带的是致病突变,其母携带的基因突变意义未明,予小剂量左旋多巴进行诊断性治疗,予口服美多芭每次0.03 g,每日3次治疗,肌张力改善效果显著,随后患儿亦伴有异动症,辅以苯海索片(每次2 mg,每日3次)、氯丙嗪(每次6 mg,每日3次)调节运动,异动症明显缓解。随后患儿运动症状渐好转,6个月后能独坐,四肢有力,可扶走,能与人简单交流。
2 讨论
DRD于1976年,由日本学者SEGAWA首先报道,亦称Segawa病[1]。DRD的发病率为0.5/100万,以女性多见,发病率女:男为2:1~4:1。DRD临床表现呈慢性进行性加重,肌张力障碍、肢体震颤、动作倒退是其主要临床症状[2],早期症状有明显的昼夜波动性,晨轻暮重,晚期昼夜波动性消失,症状严重而持久,体征可见踝阵挛阳性,腱反射亢进,肌张力增高或减低,肌张力可呈齿轮样或铅管样增高,但病理征多数阴性,主要辅助检查如脑电图、MR、CT、PET等多为阴性结果[3]。另外,DRD患者体内可能出现的泌乳素水平升高、脑脊液生物蝶蛉和新喋呤水平降低[4]以及生长激素分泌抑制[5-6]等病理变化都与多巴胺神经元功能紊乱有关。DRD有常染色体显性及隐性遗传两种方式。三磷酸鸟苷环化水解酶 1(three guanosine monophosphate cyclohydrolase I,GCH-1)基因突变与显性遗传DRD相关,基因定位于14q22.1-q22.2,多见,外显率不全,存在散发病例,临床症状较轻[7-8]。当个体体内GCH-1活性下降至正常活性的20%甚至更低时,脑内多巴胺合成量明显不足,临床上就会表现出肌张力障碍或帕金森样症状[8]。TH基因突变与隐性遗传DRD相关,基因定位于11p15.5,已报道致病基因60余种[9]。TH基因突变可直接导致TH活性降低,从而直接影响多巴胺及其下游物质的合成[10]。TH缺陷型DRD分2种类型:TH严重缺陷型,亦称新生儿脑病,因酶活性低,发病早,病情重,危及生命;TH轻中度缺陷型,多在婴儿期发病,进行性运动功能减退-强直伴肌张力障碍综合征,多见,多于1岁内起病,症状呈昼夜波动性,一般无智力损害[11-12]。
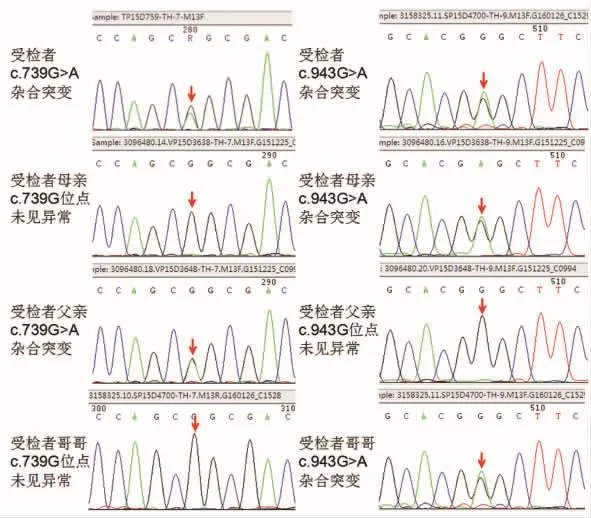
图2 例2患儿及其父母、胞兄的基因检测结果
本文2例病例均为女性患儿,婴儿期发病,为散发病例(如图3),以运动发育倒退、肌张力障碍为主要表现,症状呈明显昼夜波动性,其中1例有明显帕金森样症状,另1例呈肌张力低下,国内也有类似病例报道[13],以肌张力低下为主要表现的TH缺陷型DRD,结合2例发病年龄和临床症状,可推测这2例均属TH轻中度缺陷型。2例头颅MR、脑电图、脊肌萎缩症SMN1基因测序均未见明显异常,均以常染色体隐性遗传,TH基因杂合突变引起酪氨酸羟化酶缺乏致病,例1患儿TH基因6号外显子上c.694C>T,为致病突变,该突变为无义突变,翻译产物蛋白质第232位氨基酸由谷氨酰胺变为终止密码子,并使得蛋白质翻译提前终止,导致所编码的蛋白质发生截短从而丧失其正常功能,患儿之父携带该位点突变,患儿12号外显子上c.1240G>A,为致病突变,该突变为错义突变,使所编码的蛋白质第414位氨基酸由甘氨酸突变为精氨酸,该突变会导致酪氨酸羟化酶的活性显著降低,患儿之母携带该位点突变。例2患儿TH基因7号外显子上c.739G>A,为致病突变,该突变为错义突变,使所编码的蛋白质第247位氨基酸由甘氨酸变为丝氨酸,导致蛋白稳定性降低,患儿之父携带该位点突变,患儿9号外显子上c.943G>A,意义未明,该变异为错义突变,使所编码的蛋白质第315位氨基酸由甘氨酸变为丝氨酸,有文献报道在患者中检测到该突变,生物信息学软件预测有致病可能,患儿之母、胞兄携带该位点突变。结合例2患儿对小剂量左旋多巴治疗效果显著,从诊断性治疗角度也可确诊 DRD,因而推测c.943G>A很可能就是新发现的致病突变位点。

图3 例1与例2患儿遗传图谱
DRD对小剂量左旋多巴的治疗效果显著而持久[14]。DRD的病理变化是黑质纹状体通路结构正常,无变性改变,纹状体内多巴胺水平降低,黑质多巴胺能神经元数目正常,但细胞内色素减少,未见Lewy小体,无明显神经元退行性改变,无明显神经胶质增生,因此,DRD对小剂量多巴胺制剂治疗效果非常显著[15-16]。DRD需与注意脑性瘫痪、肝豆状核变性、少年型帕金森病以及其他类型的肌张力障碍相鉴别,根据临床表现鉴别困难时可进行左旋多巴诊断性治疗或全外显子基因测序明确诊断。多巴胺起始治疗剂量多为1 mg/(kg·d),可逐渐增加用量至出现最好控制效果,有报道,左旋多巴治疗初期易出现异动症,适当减量后异动症可明显缓解。本文2例病例,均被误诊为“脑性瘫痪”,经基因测序确诊后予小剂量美多芭治疗后运动逐渐改善,显示出显著而持久的疗效,2例患儿美多芭用量已经很少时,仍出现异动症,为保证治疗效果,经验性辅以苯海索片、氯丙嗪调节运动及肌张力,异动症明显缓解。
综上,对不明原因的运动倒退或肌张力障碍,尤其是婴儿期起病,应注意考虑THD所致DRD,可通过诊断性治疗或基因测序明确诊断。对于DRD患儿,早期确诊及治疗对其后遗症和心理疾患的防治极为重要。
[1]SEGAWA M,HOSAKA A,MIYAGAWA F,et al.Hereditary progressive dystonia with marked diurnal fluctuation[J].Adv Neurol,1976,14:215-233.
[2]SEGAWA M,NOMURA Y,NISHIYAMA N.Autosomal dominant guanosine triphosphate cyclohydrolase I deficiency(Segawa disease)[J].Ann Neurol,2003,54(6):S32-S45.
[3]BIANCA S,BIANCA M.A new deletion in autosomal dominant guanosinetriphosphate cyclohydrolase Ideficiency gene--Segawa disease[J].J Neural Transm(Vienna),2006,113(2):159-162.
[4]FURUKAWA Y,NISHI K,KONDO T,et al.CSF biopterin levels and clinical features of patients with juvenile parkinsonism [J].Adv Neurol,1993,60:562-567.
[5]FURUKAWA Y,GUTTMAN M,SPARAGANA SP,et al.Doparesponsive dystonia due to a large deletion in the GTP cyclohydrolase I gene[J].Ann Neurol,2000,47(4):517-520.
[6]SEGAWA M.Hereditary progressive dystonia with marked diurnal fluctuation[J].Brain Dev,2000,22(1):S65-S80.
[7]NYGAARD TG,WILHELMSEN KC,RISCH NJ,et al.Linkage mapping of dopa-responsive dystonia(DRD)to chromosome 14q [J].Nat Genet,1993,5(4):386-391.
[8]SEGAWA M.Hereditary progressive dystonia with marked diurnal fluctuation[J].Brain and Development,2011,33(3):195-201.
[9]刘威,唐北沙,曹贵方,等.中国人常染色体隐性遗传性多巴反应性肌张力障碍TH基因突变分析[J].中华医学遗传学杂志, 2004,21(5):36-38.
[10]FURUKAWA Y,GRAF W D,WONG H,et al.Dopa-responsive dystonia simulating spastic paraplegia due to tyrosine hydroxylase(TH)gene mutations[J].Neurology,2001,56(2):260-263.
[11]WILLEMSEN MA,VERBEEK MM,KAMSTEEG EJ,et al.Tyrosine hydroxylase deficiency:a treatable disorder of brain catecholamine biosynthesis[J].Brain,2010,133(6):1810-1822.
[12]PONS R,SYRENGELAS D,YOUROUKOS S,et al.Levodopainduced dyskinesias in tyrosine hydroxylase deficiency[J].Mov Disord,2013,28(8):1058-1063.
[13]谭冬琼,张雅芬,叶军,等.酪氨酸羟化酶缺乏症导致的多巴反应性肌张力不全一例临床特点及基因检测[J].中华儿科杂志,2014,52(8):616-619.
[14]REBOUR R,DELPORTE L,REVOL P,et al.Dopa-Responsive Dystonia andgait analysis:A case study of levodopa therapeutic effects[J].Brain Dev,2015,37(6):643-650.
[15]FURUKAWA Y,SHIMADZU M,RAJPUT AH,et al.GTP-cyclohydrolase I gene mutations in hereditary progressive amd dopa-responsive dystonia[J].Ann Neurol,1996,39(5):609-617.[16]NARDOCCI N,ZORZI G,BLAU N,et al.Neonatal dopa-responsive extrapyramidal syndrome in twins with recessive GTPCH deficiency[J].Neurology,2003,60(2):335-337.
R746.9 (
2016-11-29)
A
(责任编辑:李立)
10.3969/j.issn.1002-0152.2017.06.011
* 中山大学孙逸仙纪念医院儿科(广州 510120)
