基于简化基因组测序的大黄鱼耐高温性状全基因组关联分析
2017-08-16陈小明李佳凯王志勇蔡明夷刘贤德
陈小明 李佳凯 王志勇 蔡明夷 韩 芳 刘贤德
(集美大学水产学院, 农业部东海海水健康养殖重点实验室, 厦门 361021)
基于简化基因组测序的大黄鱼耐高温性状全基因组关联分析
陈小明 李佳凯 王志勇 蔡明夷 韩 芳 刘贤德
(集美大学水产学院, 农业部东海海水健康养殖重点实验室, 厦门 361021)
利用Illumina HiSeqTM2500测序平台, 对通过高温胁迫实验筛选得到的20尾耐高温和20尾不耐高温的大黄鱼(Larimichthys crocea)进行了简化基因组测序(SLAF-seq), 每个样本的平均测序深度达到10.26×, 共获得419211个高质量的群体单核苷酸多态性(SNP)位点 。利用TASSEL软件的混合线性模型(MLM)进行全基因组关联分析(GWAS), 共筛选到38个与大黄鱼耐高温性状显著相关的SNP位点(P<2.39E–08)。利用BLAST程序定位每个SNP位点在大黄鱼基因组中的位置, 并分析其周围的功能基因。结果在38个SNPs附近共找到26个已知的功能基因, 这些基因主要与细胞转录、代谢、免疫等功能相关。研究结果可为下一步大黄鱼耐高温分子机制解析及耐高温品种的选育提供参考。
大黄鱼; 高温胁迫; 简化基因组测序; 单核苷酸多态性; 全基因组关联分析
大黄鱼(Larimichthys crocea)是我国重要的海洋经济鱼类, 在自然海区分布于30—60 m水深, 适应温度在10—32℃, 最适生长温度在18—25℃[1,2]。当前, 大黄鱼的养殖模式仍以浅海网箱养殖为主,网箱深度为4—6 m。由于水深较浅, 夏季大黄鱼处在(或接近)其可耐受高温的时间较长, 持续高温会导致大黄鱼生长减缓、抗病力下降, 加上病原生物感染, 常常引发大黄鱼大量发病死亡。因此, 开展大黄鱼耐高温选育的研究, 对提高养殖大黄鱼的度夏成活率具有重要的参考意义。
传统的育种是依据性状的表型进行选择[3],对于耐高温品种选育, 常规的做法是先通过高温胁迫实验, 筛选出耐高温的亲鱼, 用于进一步选育[4],但这种高温胁迫实验, 常常会造成亲鱼的死亡, 即使没有死亡, 其生殖细胞也会受到损伤, 对繁殖下一代产生不良影响。随着分子生物学和基因组学的发展, 鱼类的育种研究也正在经历由传统的选择育种、杂交育种到基于基因组信息的分子育种的转变[5,6]。基于基因组的分子育种核心内容是对基因型和表型多态性的研究, 其中单核苷酸多态性(Single Nucleotide Polymorphism, SNP)因其分布广、可实现高通量检测等优点, 逐渐成为主流分子标记[7—9]。如果能够通过前期实验, 筛选出与高温相关的SNP标记, 借助分子标记对大黄鱼进行耐高温选育, 则将可以有效提高选择的准确性, 加速育种进程。
全基因组关联分析(Genome-wide association study, GWAS)旨在从全基因组范围内筛选与性状关联的SNPs[10]。GWAS分析的成本主要有2个: 第一, 用于分析的样本数量;第二, 基因分型的费用。数量越多, 其分型成本、测定性状的成本也越高。为减少成本, 只测序极端样品的GWAS方法被开发出来了, 如BSR-seq (RNA-seq based BSA)、XP-GWAS (Extreme-phenotype genome-wide association study)等。研究证明XP-GWAS可有效降低基因分型的工作量, 能够进行低成本高效益的SNP筛选[11]。SNP分型技术也逐渐由早期阶段的中、低通量, 发展到高通量的基因芯片、重测序技术[12]。其中SLAF-seq (Specific-locus amplified fragment sequencing)就是一种便宜、高效的SNP发掘与分型高通量方法[13]。
综合考虑实验成本和效率, 本研究采用SLAF-seq技术结合XP-GWAS策略在全基因组范围内寻找与大黄鱼耐高温性状相关的SNP位点, 旨在寻找新的遗传标记, 为大黄鱼耐高温性状的改良提供基础资料。
1 材料与方法
1.1 实验材料
本实验于2014年5—6月在福建省宁德市金铃水产科技有限公司育苗场进行。所用实验材料为700尾2月龄大黄鱼, 平均体重0.98 g, 平均体长4.87 cm,其亲本为宁德三都澳海区的普通养殖群体(88♀× 50♂)。实验鱼放在室内长方形水泥池(2 m×1 m×1 m)中暂养7d后开始升温实验, 暂养期间水温(25.0± 0.3)℃, 正常喂食, 每天换水一次。
1.2 实验方法
高温胁迫实验大黄鱼暂养7d后, 参照Diegane等[14]的实验方法每天升高1℃进行缓慢升温,当鱼开始死亡时, 停止升温, 维持出现死亡时的温度1d后, 再按每天升高0.5℃进行升温。在实验期间, 连续充气, 正常投喂饵料, 每天换水1次, 每2h测量水温1次, 记录大黄鱼的死亡情况, 包括每条鱼的死亡时间和死亡温度。至大黄鱼全部死亡时, 停止加热升温。在上述大黄鱼幼鱼群体热耐受性实验中, 采集最先死亡的大黄鱼20尾(定义为不耐高温个体S)与最后死亡的大黄鱼20尾(定义为耐高温个体R), 去除内脏取全鱼, 固定于95%乙醇中, –20℃保存备用。
大黄鱼基因组DNA的提取与检测利用常规的苯酚鲶氯仿鲶异戊醇法抽提DNA, 1.0%琼脂糖凝胶电泳检测基因组DNA的完整性, 紫外分光光度计测定OD260与OD280, 确定DNA的质量并将浓度调至50—100 ng/μL, 确保符合后续测序分型的要求,–20℃保存备用。
测序分型本研究利用大黄鱼基因组(Gen-Bank登录号: GCA_000742935.1)作为参考基因组,采用酶切预测软件(北京百迈客生物科技有限公司,北京)对其进行电子酶切预测。根据选定的最适酶切方案, 对各样品基因组DNA分别进行酶切。对得到的酶切片段进行5′端修复和磷酸化处理, 3′端加A, 连接solexa接头, 用琼脂糖凝胶电泳进行片段大小选择, 通过PCR扩增增大文库量, 文库质检合格后使用Illumina HiSeqTM2500测序平台测序。
统计分析利用接头序列对测序的原始数据进行分类, 得到各个样品的测序读段。去除质量得分<20的测序读段, 利用SOAP2[15]软件将合格的测序读段比对到大黄鱼参考基因组上, 将两端都比对到参考基因组上的测序读段用来定义SLAF标签。选取样本平均深度在2以上的组来定义SLAF标记。然后根据SLAF标记, 对40个样本进行SNP检测。
使用Plink (v1.07)[16]对得到的SNP进行质量控制, 弃去最小检出率低于80%和最小等位基因频率低于5%的SNP。使用TASSEL[17]软件中的混合线性模型进行SNPs和表型性状的全基因组关联分析,使用模型的公式如下: y=Xα+Qβ+e, 其中, y为表型值向量, X为SNP基因型矩阵, Q为admixture[18]生成的群体结构矩阵(K=1); α为基因型效应向量, β为固定效应向量, e为随机误差向量。采用Bonferroni方法对关联P值进行校正, 得到全基因组显著性阈值。本研究SNP标记的数目为419211个, 因此Bonferroni校正的全基因组显著性阈值为2.39E–08 (0.01/419211)。使用R (https://www.R-project.org)软件生成关联分析结果P值的Quantile-Quantile (QQ)图和曼哈顿图。
2 结果
2.1 耐高温和不耐高温大黄鱼的筛选
通过高温胁迫实验观察到大黄鱼开始出现死亡时的温度为33℃, 对高温胁迫实验中大黄鱼死亡情况按照发生死亡时的时间进行归纳整理, 最先死亡的20尾大黄鱼(定义为不耐高温个体S)和最后死亡的20尾大黄鱼(定义为耐高温个体R)的具体死亡时间见表 1。
2.2 测序结果分析
测序结果和SLAF标签统计利用大黄鱼基因组为参考基因组进行电子酶切预测, 最终确定使用RsaⅠ+HaeⅢ酶切组合, 酶切片段长度在264—294 bp的序列定义为SLAF标签, SLAF标签在基因组上基本均匀分布, 位于重复序列区的SLAF标签比例为2.52%。
对40个样品进行测序, 最终共得到98620785条测序读段, 测序平均Q30为84.71%, 平均GC含量为43.10%。使用SOAP2软件将各个样品数据比对到大黄鱼参考基因组上, 两端都比对到基因组上为可靠的测序读段, 以比对到基因组唯一位置的两端序列来做SLAF标签开发。本研究共计开发146560个SLAF标签, 每个样品的平均测序深度为 10.26×。
SNP筛选根据开发得到的146560个SLAF标记统计SNP信息, 共得到2423837个群体SNP。根据MAF>0.05和完整度>0.8进行筛选, 共得到419211个高质量群体SNP可用于后续的GWAS分析。

表 1 20尾耐高温(R)和20尾不耐高温(S)大黄鱼的死亡时间Tab.1 The information of death time of 20-tails thermal-tolerance (R) and 20-tails thermal-sensitive (S) large yellow croaker
群体结构分析通过admixture软件分析大黄鱼的群体结构, 根据交叉验证错误率的谷值确定最优分群数(K), 如图 1所示, 当K=1时, 交叉验证错误率最低, 说明本实验分析的样品应该是来自于同一个原始的祖先, 也就是样品不存在明显的分群,适合做后续的关联分析。绘制了混合线性模型下群体的QQ-plot图(图 2)。观测值与期望值前期基本相符, 在后期翘起, 说明选用的分析模型合适, 关联结果可靠。

图 1 每个K值对应的交叉验证误差Fig.1 The CV value of each K value

图 2 耐热性状的Quantile-Quantile图Fig.2 QQ-plot for thermal tolerance
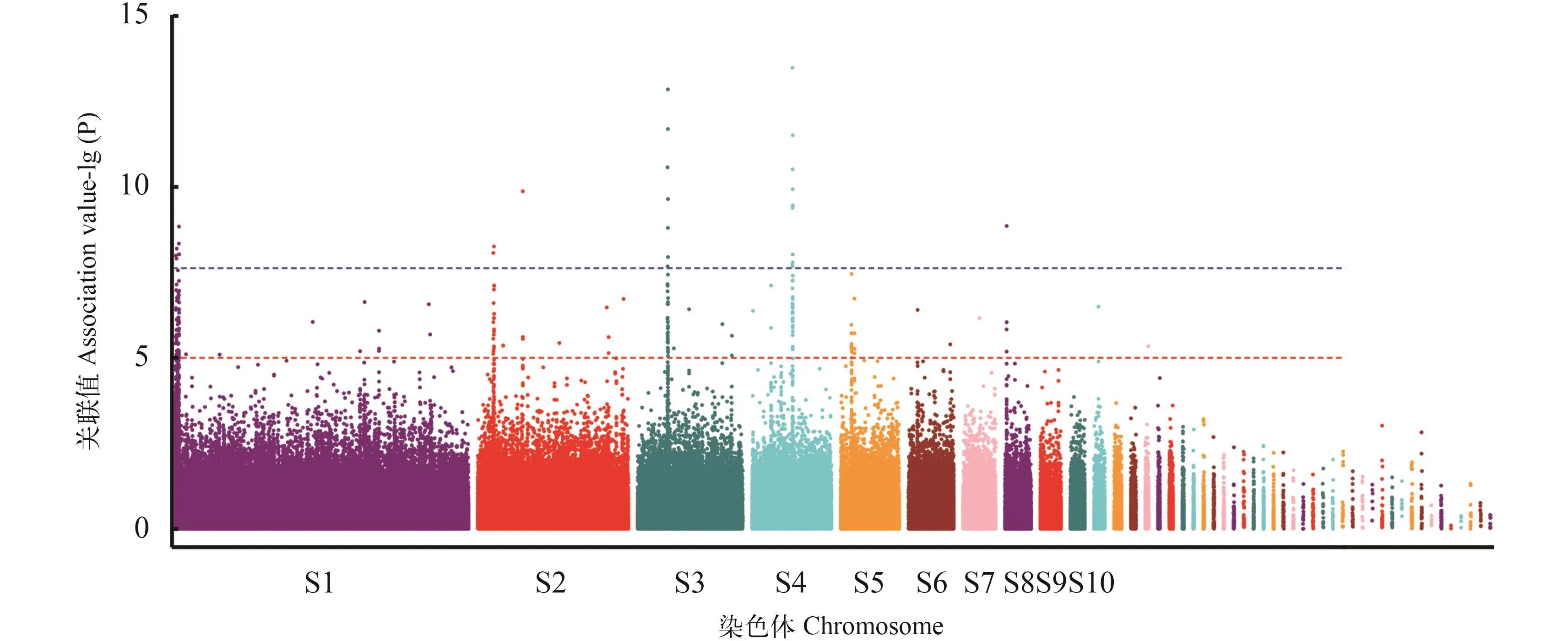
图 3 大黄鱼耐热性状全基因组关联分析的曼哈顿图Fig.3 Manhttan plot of GWAS of thermal tolerance in large yellow croaker
全基因组关联分析采用混合线性模型进行全基因组关联分析, 大黄鱼耐高温性状关联分析的曼哈顿图如图 3所示, 根据关联分析的结果, 共筛选得到38个与大黄鱼耐高温性状显著相关的SNP位点(P<2.39E–08), 其中有14个SNPs位点位于scaffold scf716上, 有10个SNPs位点位于scaffold scf2上, 有10个SNPs位点位于scaffold scf466上, 有成簇分布的特点。取SNP位点上下游各1000 bp的序列, 采用BLAST程序与大黄鱼基因组(Gen-Bank登录号: GCA_000972845.1)进行比对, E-value的阈值为1e–3, 获得每个SNP位点周围的基因,除去3个未注释到基因以及蛋白功能重复的, 共发现26个已知功能的基因, 这些基因主要与细胞转录、代谢、免疫等功能相关(表 2)。

表 2 与耐热性状显著关联的SNPs位点Tab.2 SNPs significantly associated with thermal tolerance trait
3 讨论
对于GWAS分析, 其成本主要来自于2个方面,样本数量和SNP分型方法。样本数量可以通过仅分析极端表型个体的策略来减少[11], 而SNP分型方法有很多, 有低通量(CAPS、一代测序)、中等通量(SNaPshot、质谱法)以及高通量(基因芯片、重测序)等方法[12]。目前大黄鱼尚无商业用的SNP检测芯片, 且芯片方法也只能检测已知的SNP变异。重测序方法费用比较高, 但简化基因组测序可以有效降低这个费用。目前简化基因组技术有多种方法,如RAD、GBS、2b-RAD、dd-RAD、SLAF等[13,19],各种方法均有其优势和劣势。SLAF-seq技术可以在减少测序成本的同时, 获得覆盖度较高的、能够在群体间进行分型的标记, 为非模式生物全基因组范围内的SNP标记发掘提供了高效的方法[13]。目前, 研究人员已利用该方法在玉米、黄瓜上进行全基因组关联分析研究[20,21], 在玉米上发现1个新的玉米花序分生组织突变体、在黄瓜上找到140个与白粉病抗性相关的SLAF标签, 2个热点区域。基于成本和效率的考虑, 本研究采用了前述的研究方案。
通过GWAS分析, 本研究共筛选到38个与大黄鱼耐高温性状显著相关的SNP标记(P<2.39E–08),这些SNP位点主要分布在scf2、scf466和scf716上,表现出成簇分布的特点, 这点在其他物种基因组关联分析中也有类似的结果[22,23], 可能与高温相关的这些SNP位点主要集中在部分染色体上。本研究发现的与高温相关的SNP位点涉及基因的功能主要有细胞转录、代谢、免疫等, 可能高温对大黄鱼来说是一种刺激, 引起其各种生理反应, 因而基因转录、复制以及免疫应答变得非常活跃。遗憾的是, 在本次筛查出SNP位点周围并没有找到直接与高温相关的基因, 分析其原因可能与我们所用的简化基因组测序有关, 导致一些相关的SNP位点没有筛查出来, 因而对应的基因也没有发掘出来。因此,下一步还需要用重测序的方法在大群体中进行进一步的筛查。
[1]Xue B, He Y N, Guo Y M, et al.Research of ecological culture system of Larimichthys crocea [J].Modern Agricultural Sciences and Technology, 2014, (16): 244—249 [薛彬, 何依娜, 郭远明, 等.大黄鱼生态养殖系统研究.现代农业科技, 2014, (16): 244—249]
[2]Li J K, Wang Z Y, Liu X D, et al.Effects of high temperature on serum biochemical indices of large yellow croaker Larimichthys crocea [J].Marine Science Bulletin, 2015, 34(4): 457—462 [李佳凯, 王志勇, 刘贤德, 等.高温对大黄鱼(Larimichthys crocea)幼鱼血清生化指标的影响.海洋通报, 2015, 34(4): 457—462]
[3]Hulata G.Genetic manipulations in aquaculture: a review of stock improvement by classical and modern technologies [J].Genetica, 2001, 111(1—3): 155—173
[4]Ma A J, Huang Z H, Wang X A, et al.The selective breeding of thermal tolerance family and appraisal of performance in turbot Scophthalmus maximus [J].Oceanologia et Limnologia Sinica, 2012, 43(4): 797—804 [马爱军, 黄智慧, 王新安, 等.大菱鲆(Scophthalmus maximus)耐高温品系选育及耐温性能评估.海洋与湖沼, 2012, 43(4): 797—804]
[5]Xu K, Duan W, Xiao J, et al.Development and application of biological technologies in fish genetic breeding [J].Science China Life Sciences, 2015, 58(2): 187—201
[6]Yue G H.Recent advances of genome mapping and marker-assisted selection in aquaculture [J].Fish and Fisheries, 2014, 15(3): 376—396
[7]Vignal A, Milan D, SanCristobal M, et al.A review on SNP and other types of molecular markers and their use in animal genetics [J].Genetics Selection Evolution, 2002, 34(3): 275—306
[8]Liu Z J, Cordes J F.DNA marker technologies and their applications in aquaculture genetics [J].Aquaculture, 2004, 238(1): 1—37
[9]Quan Y C, Ma D M, Bai J J, et al.SNPs identification in RNA-seq data of largemouth bass (Micropterus salmoides) fed on formulated feed and association analysis with growth trait [J].Acta Hydrobiologica Sinica, 2016, 40(6): 1128—1134 [全迎春, 马冬梅, 白俊杰, 等.大口黑鲈转录组SNPs筛选及其与生长的关联分析.水生生物学报, 2016, 40(6): 1128—1134]
[10]Korte A, Farlow A.The advantages and limitations of trait analysis with GWAS: a review [J].Plant Methods, 2013, 9(1): 29
[11]Yang J, Jiang H, Yeh C T, et al.Extreme-phenotype genome-wide association study (XP-GWAS): a method for identifying trait-associated variants by sequencing pools of individuals selected from a diversity panel [J].The Plant Journal, 2015, 84(3): 587—596
[12]Zhao Q Y, Li X, Zhou D G, et al.SNP genotyping methods for crops in post-genomic era [J].Molecular Plant Breeding, 2010, 8(1): 125—133 [赵琼一, 李信, 周德贵,等.后基因组时代下作物的SNP分型方法.分子植物育种, 2010, 8(1): 125—133]
[13]Sun X, Liu D, Zhang X, et al.SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high throughput sequencing [J].PLoS One, 2013, 8(3): e58700, doi: 10.1371/journal.pone.0058700
[14]Diegane N D, Chen Y Y, Lin Y H, et al.The immune response of tilapia Oreochromis mossambicus and its susceptibility to Streptococcus iniae under stress in low andhigh temperatures [J].Fish & Shellfish Immunology, 2007, 22(6): 686—694
[15]Li R, Yu C, Li Y, et al.SOAP2: an improved ultrafast tool for short read alignment [J].Bioinformatics, 2009, 25(15): 1966—1967
[16]Purcell S, Neale B, Todd-Brown K, et al.PLINK: a tool set for whole-genome association and population-based linkage analyses [J].The American Journal of Human Genetics, 2007, 81(3): 559—575
[17]Bradbury P J, Zhang Z, Kroon D E, et al.TASSEL: software for association mapping of complex traits in diverse samples [J].Bioinformatics, 2007, 23(19): 2633—2635
[18]Alexander D H, Novembre J, Lange K.Fast model-based estimation of ancestry in unrelated individuals [J].Genome Research, 2009, 19(9): 1655—1664
[19]Davey J W, Hohenlohe P A, Etter P D, et al.Genomewide genetic marker discovery and genotyping using next-generation sequencing [J].Nature Reviews Genetics, 2011, 12(7): 499—510
[20]Xia C, Chen L, Rong T, et al.Identification of a new maize inflorescence meristem mutant and association analysis using SLAF-seq method [J].Euphytica, 2015, 202(1): 35—44
[21]Zhang P, Zhu Y, Wang L, et al.Mining candidate genes associated with powdery mildew resistance in cucumber via super-BSA by specific length amplified fragment (SLAF) sequencing [J].BMC Genomics, 2015, 16: 1058, doi: 10.1186/s12864-015-2041-z
[22]Li H, Peng Z, Yang X, et al.Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels [J].Nature Genetics, 2013, 45(1): 43—50
[23]Huang X, Wei X, Sang T, et al.Genome-wide association studies of 14 agronomic traits in rice landraces [J].Nature Genetics, 2010, 42(11): 961—967
GENOME-WIDE ASSOCIATION STUDY OF THERMAL TOLERANCE IN LARGE YELLOW CROAKER LARIMICHTHYS CROCEA BASED ON SLAF-SEQ TECHNOLOGY
CHEN Xiao-Ming, LI Jia-Kai, WANG Zhi-Yong, CAI Ming-Yi, HAN Fang and LIU Xian-De
(Key Laboratory of Mariculture for the East China Sea, Ministry of Agriculture; Fisheries College, Jimei University, Xiamen 361021, China)
Twenty thermal-tolerant and twenty thermal-sensitive individuals of Larimichthys crocea were sequenced using specific-locus amplified fragment (SLAF-seq) technology based on Illumina HiSeqTM2500 platform.419211 SNPs were identified with an average read depth of 10.26× for each sample.Thirty-eight SNPs (P<2.39E–08) significantly related with thermal tolerance trait were identified according to association analysis.The SNP locations in large yellow croaker genome were identified using BLAST program, and functional genes around SNP were annotated.Twenty-six genes with known functions were discovered around 38 SNPs, which mainly regulate cell transcription, metabolism and immunity.These results provide basic information to analyze thermal-tolerant molecular mechanism and develop thermal-tolerant lines of Larimichthys crocea in the future.
Larimichthys crocea; High temperature; SLAF-seq; SNP marker; GWAS
Q344+.1
A
1000-3207(2017)04-0735-06
10.7541/2017.91
2016-06-22;
2016-11-05
国家自然科学基金(31172397和31402339); 福建省高等学校新世纪优秀人才支持计划(JA14167)资助 [Supported by the National Natural Science Foundation of China (31172397, 31402339); the New Century Excellent Talents of Fujian Province University (JA14167)]
陈小明(1991—), 男, 江西南康人; 硕士研究生; 主要从事水产生物遗传育种研究。E-mail: x_m_chen@163.com
刘贤德, 教授; 主要从事水产生物遗传育种研究。E-mail: xdliu@jmu.edu.cn
