肢体缺血再灌注后肺损伤小鼠肺组织AT1R和Mas受体蛋白表达变化*
2017-08-07王枭鹰SHAHINAhmedMUHAMMEDRafi李树民杨秀红
刘 帆, 王枭鹰, SHAHIN Ahmed, MUHAMMED Rafi-N, 李树民, 杨秀红
(华北理工大学基础医学院生理学系, 河北省慢性疾病重点实验室, 唐山市慢性病临床基础研究重点实验室, 河北 唐山 063000)
肢体缺血再灌注后肺损伤小鼠肺组织AT1R和Mas受体蛋白表达变化*
刘 帆, 王枭鹰, SHAHIN Ahmed, MUHAMMED Rafi-N, 李树民, 杨秀红△
(华北理工大学基础医学院生理学系, 河北省慢性疾病重点实验室, 唐山市慢性病临床基础研究重点实验室, 河北 唐山 063000)
目的: 通过观察小鼠肢体缺血再灌注(LIR)后不同时点肺组织血管紧张素Ⅱ 1型受体(AT1R)和Mas受体蛋白表达与肺损伤的变化,探讨局部组织AT1R和Mas受体蛋白表达失衡在LIR急性肺损伤(ALI)中的作用。方法:42只8周龄雄性ICR小鼠随机分为7组,每组6只,其中1组作为对照组,其余6组为再灌注0.5 h、1 h、2 h、4 h、6 h和12 h模型组。模型组小鼠用橡皮圈结扎双后肢根部,缺血2 h后剪断橡皮圈,分别于再灌注后不同时点眼球取血处死小鼠。取肺组织计算脏器系数和湿/干重比;肺泡灌洗液细胞计数和蛋白浓度检测;肺组织病理切片常规HE染色观察肺组织形态变化并进行病理损伤评分;Western blot检测肺组织AT1R和Mas受体蛋白的表达。结果:模型组小鼠肺脏器系数、湿/干重比、肺泡灌洗液细胞计数和蛋白浓度在LIR后显著升高。病理学结果显示,LIR后不同时点小鼠肺组织出现肺泡壁毛细血管扩张和充血、间质和肺泡水肿、血管壁和支气管壁炎症细胞浸润、肺泡间隔增厚、炎症细胞浸润及肺气肿等不同程度的损伤变化,且随着再灌注时间的延长,肺损伤评分逐渐升高。Western blot结果显示,AT1R蛋白在再灌注0.5 h时开始升高,1 h达到最高,之后随再灌注时间的延长,AT1R表达逐渐降低;Mas受体蛋白随再灌注时间延长逐渐升高。结论:LIR引起急性肺损伤,并随再灌注时间的延长损伤逐渐加重;AT1R和Mas受体蛋白表达的变化可能与小鼠LIR后急性肺损伤有关。
缺血再灌注; 肺损伤; 肾素-血管紧张素系统; 血管紧张素Ⅱ 1型受体; Mas受体
肢体缺血再灌注(limb ischemia-reperfusion,LIR)可引发急性肺损伤(acute lung injury,ALI)。大量研究证实,肺组织局部肾素-血管紧张素系统(renin-angiotensin system,RAS)稳态失衡与急性肺损伤的发生发展有关。RAS是体内重要的体液调节系统,包括血管紧张素转换酶(angiotensin-converting enzyme,ACE)-血管紧张素Ⅱ(angiotensin Ⅱ,AngⅡ)-血管紧张素Ⅱ 1型受体(angiotensinⅡ type 1 receptor,AT1R)与血管紧张素转换酶2(angiotensin-converting enzyme 2,ACE2)-血管紧张素(1-7)[angiotensin (1-7),Ang (1-7)]-Mas受体2条轴,以循环RAS和局部RAS两种方式发挥作用。ACE的效应分子AngⅡ通过与AT1R结合,发挥调节血压和水钠平衡,促炎症、促增殖和收缩血管等作用。ACE2的效应分子Ang (1-7)通过与Mas受体结合,发挥拮抗ACE-AngⅡ-AT1R 轴的作用。文献研究已证实,在急性呼吸窘迫综合征(acute respiratory distress syndrome,ARDS)病人和动物模型,ACE活性升高,ACE2活性下降,ACE2-Ang (1-7)-Mas轴在AngⅡ/AT1R介导的肺损伤中发挥了重要的作用[1-2]。
我们前期研究发现,LIR小鼠肺组织ACE表达升高,ACE2表达降低;肺组织Ang II和Ang (1-7)表达均升高,再灌注早期Ang (1-7)高于Ang II,晚期Ang (1-7)低于Ang II;而再灌注后循环Ang (1-7)逐渐下降,Ang II逐渐升高;这些变化在ACE2 基因敲除的LIR小鼠加重,在ACE2转基因LIR小鼠减轻[3]。结合文献结果,我们推测,ACE-AngⅡ-AT1 轴和ACE2-Ang(1-7)-Mas轴的作用失衡,可能在再灌注导致的急性肺损伤中发挥重要作用。
本研究采用前期成熟方法复制再灌注急性肺损伤动物模型,观察LIR小鼠再灌注后不同时点,肺组织AT1R和Mas受体蛋白的表达与肺损伤的变化,进一步探讨ACE-AngⅡ-AT1 轴和ACE2-Ang (1-7)-Mas轴在LIR所致ALI中的作用。
材 料 和 方 法
1 实验动物和主要试剂
随机选取雄性8周龄野生型ICR小鼠42只,体重 25~35 g,由中国医学科学院实验动物研究所提供,在华北理工大学实验动物中心SPF级动物房进行饲养,自由进食水。
AT1R抗体购自Santa Cruz;Mas受体抗体购自Alomone Labs;β-actin抗体购自杭州华安生物技术有限公司;RIPA组织蛋白裂解液、BCA蛋白浓度检测试剂、彩色预染蛋白标准分子量、Western blot用II 抗、ECL发光剂、醋酸纤维素膜、Western blot抗体稀释液均购自碧云天生物技术研究所。
2 方法
2.1 动物分组 雄性8周龄野生型ICR小鼠42只随机分为7组,每组6只。其中1组作为对照组,其余6组为模型组,分别为再灌后0.5 h、1 h、2 h、4 h、6 h和12 h组,对照组不结扎双侧后肢,其余操作同模型组。
2.2 复制小鼠肢体缺血再灌注模型的建立 采用我室常规方法复制小鼠肢体缺血再灌注模型[3]。腹腔注射水合氯醛(3 mg/kg)麻醉小鼠,在双后肢根部行橡皮圈结扎术,缺血2 h后剪断橡皮圈,按摩双后肢恢复血液灌注。分别于再灌注后0.5 h、1 h、2 h、4 h、6 h和12 h摘除眼球,取血处死小鼠,留取肺组织,-80 ℃保存。
2.3 病理组织学方法检测肺损伤 将一侧肺浸泡于4%甲醛中固定,常规方法制作病理组织切片,光镜下观察肺组织病理变化。病变分为4种类型:肺泡壁毛细血管扩张充血;肺泡出血;肺泡壁和血管壁嗜中性粒细胞浸润; 肺泡壁增厚。按照病变的严重程度分为5个等级:0-无损伤;1-轻度损伤(每个视野有≤25%损伤);2-中度损伤(每个视野有26%~50%损伤);3-重度损伤(每个视野有51%~75%损伤);4-极重度损伤(≥76%弥漫性肺损伤,伴有大量炎细胞浸润)。计算视野中4种病变损伤程度的得分总和; 10个随机视野得分的平均值为每组最终得分[3]。
2.4 肺脏器系数和干湿质量比测定 取小鼠双肺组织,滤纸吸干肺组织表面血液,采用分析天平称量双肺重量,电子天平测量小鼠体重,脏器系数=脏器重量/体重×100%;取小鼠右肺上叶,滤纸吸干肺组织表面血液,采用分析天平称湿重后,置于电热真空干燥箱内,60 ℃烘烤72 h,称干重,计算湿/干重比(wet/dry weight ratio)。
2.5 支气管肺泡灌洗液(bronchoalveolar lavage fluid,BALF)细胞计数和蛋白浓度检测 小鼠仰卧位固定后,行颈部正中切口,分离气管,插入气管导管,固定,以1 mL预冷的PBS行肺泡灌洗,停留2 min后收集灌洗液,反复灌洗3次,4 ℃下1 500 r/min离心10 min,收集上清液。血细胞计数板计数细胞总数;BCA法检测蛋白浓度。
2.6 Western blot检测肺组织AT1R和Mas受体蛋白表达 每组称取100 mg肺组织,加入1 mL蛋白裂解液RIPA,冰上超声匀浆,反复3次;4 ℃下12 000 r/min离心15 min,取上清分装。采用BCA 法测定上清液中的蛋白总浓度,并用裂解液调整蛋白浓度至6 g/L,与5×上样缓冲液按4∶1混匀,煮沸10 min;进行SDS-PAGE,硝酸纤维素膜转膜,5%脱脂奶粉封闭 1 h, I 抗4 ℃孵育过夜(AT1R抗体1∶300,Mas受体抗体1∶300,β-actin 抗体1∶1 000);次日用TBST洗膜3次,每次10 min; II 抗37 ℃孵育1 h,TBST洗膜3次,每次10 min;ECL显影、成像。利用Image Lab软件对结果进行相对定量分析。
3 统计学处理
采用SPSS 17.0软件进行统计学分析,所有结果以均数±标准差(mean±SD)表示。多组间均数的比较采用单因素方差分析(one-way ANOVA),两两比较采用Dunnett’s T3检验。以P<0.05为差异有统计学意义。
结 果
1 肺脏器系数和湿/干重比
肺脏器系数和肺湿/干重比实验结果见图1。与对照组相比,缺血2 h,再灌注1 h、2 h、4 h、6 h和12 h模型组小鼠肺脏器系数均升高,以4 h组升高最为明显,差异有统计学意义(P<0.05);与对照组相比,再灌注0.5 h、1 h、2 h、4 h、6 h和12 h模型组小鼠肺湿/干重比也出现升高,以4 h组升高最为明显,差异有统计学意义(P<0.01),且与肺脏器系数变化趋势一致。
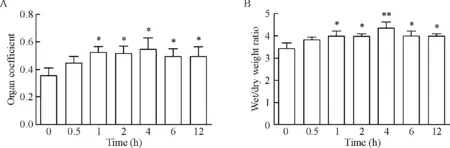
Figure 1.The organ coefficient (A) and wet/dry weight ratio (B) of lung tissue of LIR mice at different time points. Mean±SD.n=6.*P<0.05,**P<0.01vs0 h.
图1 缺血再灌注不同时点小鼠肺脏器系数和湿/干重比
2 支气管肺泡灌洗液细胞计数和蛋白浓度
LIR后支气管肺泡灌洗液细胞计数和蛋白浓度见图2。与对照组相比,再灌注0.5 h,细胞总数开始升高,2 h显著升高(P<0.05),4 h达到最高(P<0.01),此后逐渐下降;肺泡灌洗液中蛋白浓度,依灌注时间的延长逐渐升高(P<0.05),至再灌注4 h升高更为显著(P<0.01)。

Figure 2.The cell number (A) and protein concentration (B) in BALF of LIR mice at different time points. Mean±SD.n=6.*P<0.05,**P<0.01vs0 h.
图2 缺血再灌注不同时点小鼠支气管肺泡灌洗液细胞计数和蛋白浓度
3 肺组织病理变化及肺损伤评分
肉眼观模型组小鼠肺组织体积增大,呈暗红色。光镜下观察:对照组肺泡结构清晰,肺泡壁完整,间质无充血、水肿和炎症细胞浸润;模型组出现以下病理变化:肺泡壁毛细血管扩张,充血;肺泡渗出、血管壁和支气管壁炎症细胞浸润、间质水肿;部分肺泡间隔断裂,肺泡融合,形成肺气肿;肺泡间隔增厚伴炎症细胞浸润。
与对照组相比,LIR 0.5 h肺泡壁毛细血管扩张充血,血管壁和支气管壁炎症细胞浸润,损伤评分升高(P<0.01);随着再灌注时间的延长,肺组织损伤逐渐加重。镜下可见肺泡间隔明显增厚,大量炎症细胞浸润、肺组织边缘肺气肿等病理改变,肺组织损伤评分逐步升高,各模型组与对照组相比均有统计学意义(P<0.01)。而LIR 6 h和12 h组渗出改变较4 h组有所好转,病理改变以炎症细胞浸润和肺泡间隔增厚为主,见图3。
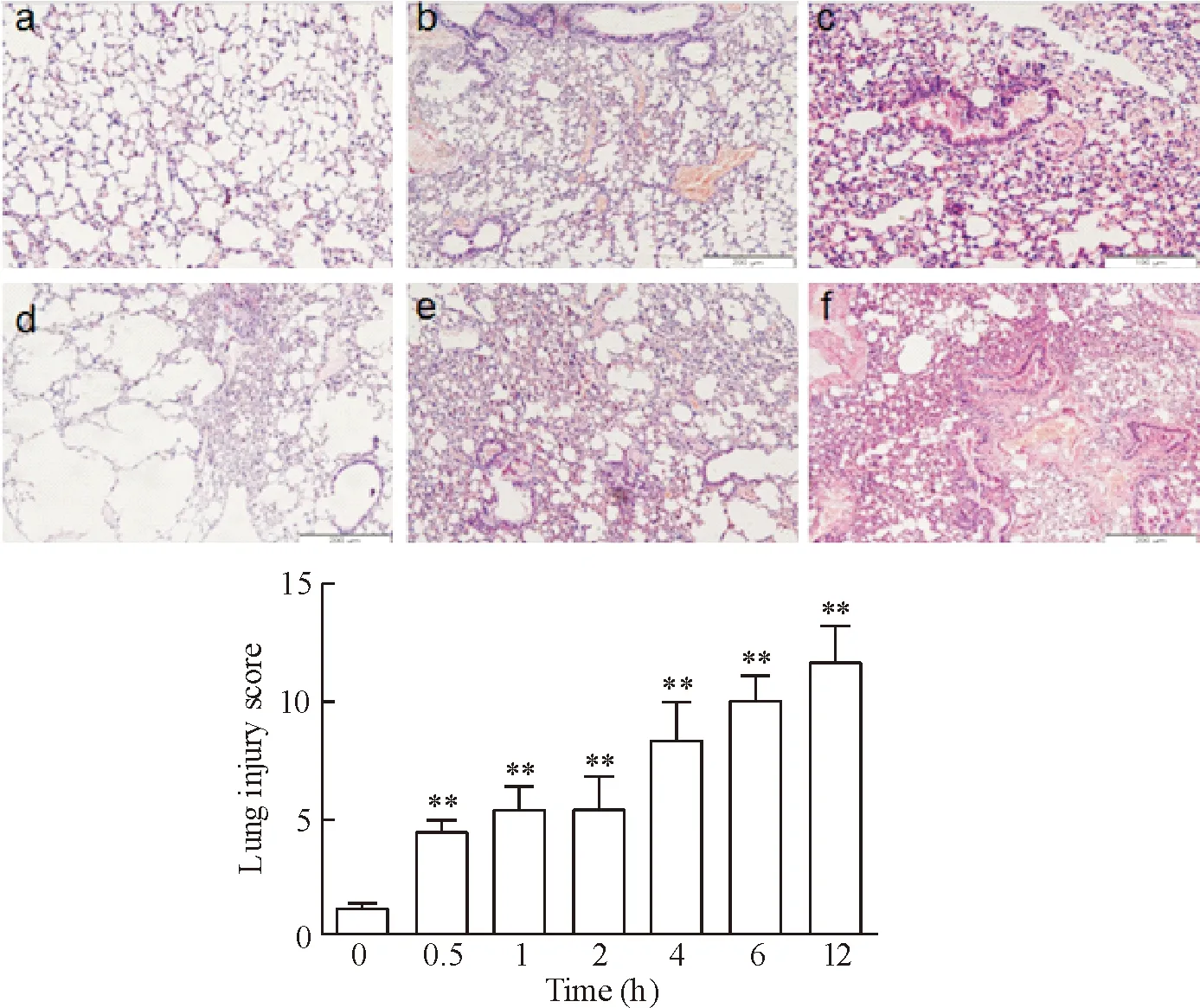
Figure 3.The lung pathological damages (HE staining, ×200) and lung injury scores of the LIR mice at different time points. a: normal lung tissue; b: telangiectasia and congestion; c: inflammatory cell infiltration around vascular and bronchi, tracheal hemorrhage; d: emphysema; e, f: thickened alveolar septa and inflammatory cell infiltration. Mean±SD.n=6.**P<0.01vs0 h.
图3 缺血再灌注不同时点小鼠肺损伤的病理变化和肺组织损伤的评分
4 局部肺组织AT1R和Mas受体蛋白表达
与对照组相比,AT1R蛋白在缺血2 h再灌注0.5 h时开始升高(P<0.05),1 h达到最高(P<0.01);之后,随灌注时间的延长,AT1R表达总体趋势逐渐降低。与之相应的Mas受体蛋白表达出现了不同的变化。与对照组相比,Mas受体蛋白随灌注时间延长逐渐升高,4 h开始显著升高,12 h达到峰值(P<0.01)。计算再灌注不同时点小鼠肺组织AT1R与Mas受体蛋白表达的比值,可见AT1R/Mas受体自再灌注1 h逐渐降低,4 h开始显著降低(P<0.01),见图4。
讨 论
LIR是一种临床常见的病理生理过程,多见于断肢再植、四肢大血管栓塞或损伤恢复血流及长时间应用止血带等情况。再灌注损伤不仅存在于缺血组织,尚可累及肺脏、肝脏、肾脏等远隔器官。肺脏是最常受累的器官之一,但确切的机制尚未完全明了。近年来的研究证实,循环和肺组织局部RAS稳态失衡与肺部疾病的发生发展有关。研究显示,高氧诱导的肺纤维化大鼠,ACE、Ang II和AT1受体显著上调[4],肾素转基因小鼠对脂多糖更加敏感,所致肺损伤也更为严重[5]。内毒素休克家兔肺局部Ang II持续升高,氧合指数下降,肺功能受损[6]。盲肠结扎穿孔术小鼠RAS系统激活,小鼠出现急性肺损伤,肺组织W/D明显增加,血氧含量下降及明显的肺损伤表现[7]。这些结果说明RAS的过度激活参与了肺损伤的发生。我们前期研究发现,LIR小鼠肺组织ACE/ACE2和Ang II/Ang (1-7)表达失衡在LIR诱发的ALI中具有重要作用[3]。本研究在此基础上,观察了LIR后肺组织RAS两条作用轴在受体水平(AT1R和Mas受体)蛋白表达的变化,进一步探讨RAS在LIR急性肺损伤中的作用。
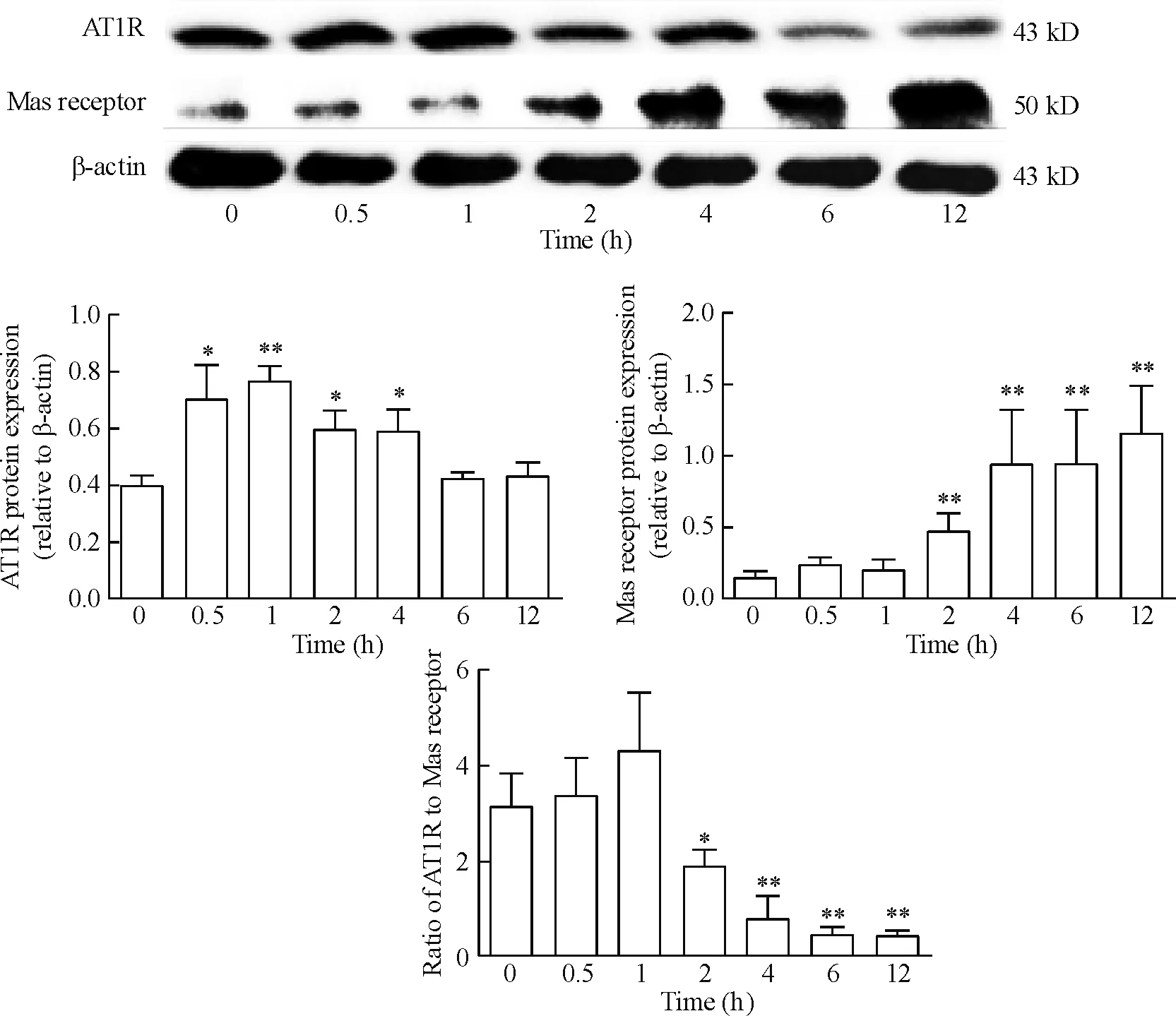
Figure 4.The protein expression of AT1R and Mas receptor in the lung tissues of LIR mice at different time points. Mean±SD.n=6.*P<0.05,**P<0.01vs0 h.
图4 缺血再灌注不同时点小鼠肺组织AT1R和Mas受体蛋白的表达
Ang II是一种重要的血管损伤因子,可通过AT1R介导引起内皮细胞肿胀、变性和凋亡,在多种器官损伤中发挥作用。Ang II与AT1R结合,可通过上调NADPH氧化酶活性,促进白细胞-内皮相互作用,进而引起ALI早期肺血管内皮细胞损伤[8],降低肺泡液体清除率[9-11],增加肺泡毛细血管壁通透性。此外,Ang II通过AT1R,显著诱导肺泡上皮细胞凋亡,导致上皮屏障功能受损、通透性增加[11]。因此,Ang II对肺泡上皮细胞和血管内皮细胞的损伤均导致肺泡渗出液和蛋白质含量的增加。研究显示,在脂多糖诱导的大鼠急性肺损伤中,肺组织Ang II和AT1R显著升高[12]; AT1R阻断剂则可抑制肺上皮细胞[12-13]和血管内皮细胞[14]凋亡,减轻肺损伤程度。我们的研究结果显示,AT1R蛋白在LIR后0.5 h和1 h增加;且肺组织出现充血、水肿, W/D比值和脏器系数增大,BALF细胞总数和蛋白含量增加。结合前期研究结果:LIR后肺组织ACE和Ang II表达上调[3],提示LIR早期ACE表达增加,提高了其效应分子Ang II的水平,高水平的Ang II可能通过AT1R促进ALI的发生。
Mas受体是Ang (1-7)发挥作用的主要受体。体内外实验表明,Ang (1-7)与Mas受体结合后,可通过阻断相关信号转导通路,发挥抑制肺成纤维细胞增殖、迁移,抑制炎症反应,防止肺泡上皮细胞凋亡等作用[15-18]。Ang (1-7)可减轻LPS刺激诱导的肺组织炎症、水肿和肺纤维化,显著降低肺损伤评分,改善肺功能,增加血氧饱和度[1, 15]。Sampaio等[19]证实Ang (1-7)通过Mas受体激活内皮型一氧化氮合酶,调节血管内皮功能。Ang (1-7)表达减少时,肺组织炎症细胞因子增加,胶原过度沉积。Mas受体激动剂的应用可减轻大鼠损伤肺组织中性粒细胞浸润,预防肺动脉高压和右心室肥大的发生[20]。靶向抑制Mas基因的表达,Ang (1-7)的作用明显下降[21]。我们的结果显示,LIR早期肺组织Mas受体蛋白表达水平较低。因此我们推测,Ang (1-7)[3]和Mas受体蛋白的低表达,也是肺损伤发生的原因之一。
Ang (1-7)激活Mas受体,使其表达增加[22];同时,Ang (1-7)还是内源性AT1R拮抗剂,可以与AT1R竞争性结合,下调AT1R[23]。虽然本研究中LIR早期AT1R蛋白表达水平暂时升高,但此时肺组织Ang (1-7)高于Ang II[3],高水平的Ang (1-7) 一方面使Mas受体表达逐渐增加,另一方面使AT1受体表达逐渐降低,二者呈现相反的变化趋势,与文献研究结果一致[22-23]。此外,Mas受体也是AT1R的生理性拮抗剂,它可能通过改变AT1R的转运特性,降低AT1R的表达[24]。结合本研究结果,我们认为,AT1R和Mas受体蛋白差异性表达参与了LIR急性肺损伤。
如前所述,Ang (1-7)和Mas受体对肺损伤发挥保护作用,而研究结果却显示肺损伤程度逐渐加重,推测有2种原因可以解释:一是AT1R和Mas受体之间存在相互作用,它们通过跨膜区域的疏水作用和分子间二硫键形成异源二聚体[24-25]。AT1R和Mas受体的共表达,使其与AngⅡ结合的能力增加了135%[24],此时肺组织Ang II也较高[3],因而肺损伤继续加重。虽然Ang (1-7)和Mas受体的表达逐渐升高,但还不足以对抗AngⅡ和AT1受体的作用。另外一个原因是,我们只观察了缺血再灌注后12 h的肺损伤变化,观察时间相对较短;Chen等[15]报道,脂多糖诱导的肺损伤变化在损伤后7 d有所恢复,而外源性给予Ang (1-7)干预,在损伤后3 d才能通过Mas受体发挥保护作用。虽然如此,研究结果已显示出Ang (1-7)和Mas受体逐渐增加的趋势,并在4 h开始明显升高。这种升高虽不能逆转肺损伤的进展,但从病理组织学上部分改善了肺泡壁和毛细血管壁的通透性,减少了渗出。因此,再灌注6 h和12 h的肺组织脏器系数、W/D、BALF细胞总数和蛋白含量较4 h略有下降(除BALF细胞总数外)。此外,类似的研究结果在课题组对LIR肾损伤中AT1R和Mas受体蛋白表达的研究中也得到证实[26]。
综合大量文献及本研究结果,缺血再灌注后局部肺组织AT1R蛋白和Mas受体蛋白的差异性表达可能参与了急性肺损伤的发生与发展,但其与急性肺损伤发生的关系及相关机制,还需进一步深入研究。
[1] Wösten-van Asperen RM, Lutter R, Specht PA, et al. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist[J]. J Pathol, 2011, 225(4):618-627.
[2] Wösten-van Asperen RM, Bos AP, Bem RA, et al. Imbalance between pulmonary angiotensin-converting enzyme and angiotensin-converting enzyme 2 activity in acute respiratory distress syndrome[J]. Pediatr Crit Care Med, 2013, 14(9):e438-e441.
[3] Chen LN, Yang XH, Nissen DH, et al. Dysregulated renin-angitotensin system contributes to acute lung injury caused by hind-limb ischemia-reperfusion in mice[J]. Shock, 2013, 40(5):420-429.
[4] Jiang JS, Lang YD, Chou HC, et al. Activation of the renin-angiotensin system in hyperoxia-induced lung fibrosis in neonatal rats[J]. Neonatology, 2012, 101(1):47-54.
[5] Wang J, Chen L, Chen B, et al. Chronic activation of the renin-angiotensin system induces lung fibrosis[J]. Sci Rep, 2015, 5:15561.
[6] 陆国平, 邬惊雷, 陆铸今, 等. 内毒素休克家兔心、肺组织局部血管紧张素系统变化[J]. 小儿急救医学, 2005, 12(5):389-391.
[7] 沈利汉, 莫红缨, 蔡立华, 等. 肾素-血管紧张素系统与ALI/ARDS的关系探讨[J]. 中国呼吸与危重监护杂志, 2010, 9(3):310-314.
[8] Deng J, Wang DX, Deng W, et al. Regulation of alveolar fluid clearance and ENaC expression in lung by exogenous angiotensin II[J]. Respir Physiol Neurobiol, 2012, 181(1):53-61.
[9] 曹春水, 殷 勤, 黄 亮, 等.血管紧张素Ⅱ对急性肺损伤大鼠肺水通道蛋白1表达的影响[J].中国危重病急救医学, 2010, 22(7):426-429.
[10]Deng J, Wang DX, Deng W, et al. The effect of endogenous angiotensin II on alveolar fluid clearance in rats with acute lung injury[J]. Can Respir J, 2012, 19(5):311-318.
[11]Lee YH, Mungunsukh O, Tutino RL, et al.Angiotensin-II-induced apoptosis requires regulation of nucleolin and Bcl-xLby SHP-2 in primary lung endothelial cells[J]. J Cell Sci, 2010, 123(Pt 10):1634-1643.
[12]Liu L, Qiu HB, Yang Y, et al. Losartan, an antagonist of AT1receptor for angiotensin II, attenuates lipopolysaccharide-induced acute lung injury in rat[J]. Arch Biochem Biophys, 2009, 481(1):131-136.
[13]Chen FP, Gong LK, Zhang L, et al.Early lung injury contributes to lung fibrosis via AT1 receptor in rats[J]. Acta Pharmacol Sin, 2007, 28(2):227-237.
[14]Akishita M, Nagai K, Xi H, et al. Renin-angiotensin system modulates oxidative stress-induced endothelial cell apoptosis in rats[J]. Hypertension, 2005,45(6): 1188-1193.
[15]Chen Q, Yang Y, Huang Y, et al. Angiotensin-(1-7) attenuates lung fibrosis by way of Mas receptor in acute lung injury[J]. J Surg Res, 2013, 185(2):740-747.
[16]Meng Y, Yu CH, Li W, et al. Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis protects against lung fibrosis by inhibiting the MAPK/NF-κB pathway[J]. Am J Respir Cell Mol Biol, 2014, 50(4):723-736.
[17]Meng Y, Li T, Zhou G, et al. The angiotensin-converting enzyme 2/angiotensin (1-7)/Mas axis protects against lung fibroblast migration and lung fibrosis by inhibiting the NOX4-derived ROS-mediated RhoA/Rho kinase pathway[J]. Antioxid Redox Signal, 2015, 22(3):241-258.
[18]El-Hashim AZ, Renno WM, Raghupathy R, et al. Angiotensin-(1-7) inhibits allergic inflammation, via the MAS1 receptor, through suppression of ERK1/2- and NF-κB-dependent pathways[J]. Br J Pharmacol, 2012, 166(6):1964-1976.
[19]Sampaio WO,Souza dos Santos RA,Faria-Silva R,et al.Angiotensin-(1-7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways[J]. Hypertension, 2007, 49(1):185-192.
[20]Wagenaar GT, Laghmani El H, Fidder M, et al. Agonists of MAS oncogene and angiotensin II type 2 receptors attenuate cardiopulmonary disease in rats with neonatal hyperoxia-induced lung injury[J]. Am J Physiol Lung Cell Mol Physiol, 2013, 305(5):L341-L351.
[21]范珊珊,贺红焰,夏纪毅,等.Mas基因沉默对血管紧张素-(1-7)拮抗血管紧张素Ⅱ诱导的大鼠肾间质成纤维细胞活化的影响[J].中国病理生理杂志, 2013, 29(7):1225-1229.
[23]Stegbauer J, Vonend O, Oberhauser V, et al. Effects of angiotensin-(1-7) and other bioactive components of the rennin-angiotensin system on vascular resistance and noradrenaline release in rat kidney[J]. J Hypertens, 2003, 21(7):1391-1399.
[24]Kostenis E, Milligan G, Christopoulos A, et al. G-protein-coupled receptor Mas is a physiological antagonist of the angiotensin II type 1 receptor[J]. Circulation, 2005, 111(14):1806-1813.
[25]Salahpour A, Angers S, Bouvier M. Functional significance of oligomerization of G-protein-coupled receptors[J]. Trends Endocrinol Metab, 2000, 11(5):163-168.
[26]王建辉, 张 伟, 刘 燕, 等. 小鼠肢体缺血再灌注后肾组织AT1和Mas受体蛋白差异性表达与肾损伤的关系[J]. 解放军医学杂志, 2016, 41(3):184-188.
(责任编辑: 林白霜, 罗 森)
Changes of lung AT1R and Mas receptor protein expression in lung tissue of mice with lung injury after limb ischemia-reperfusion
LIU Fan, WANG Xiao-ying, SHAHIN Ahmed, MUHAMMED Rafi-N, LI Shu-min, YANG Xiu-hong
(DepartmentofPhysiology,SchoolofBasicMedicalSciences,NorthChinaUniversityofScienceandTechnology,HebeiKeyLaboratoryforChronicDiseases,TangshanKeyLaboratoryforPreclinicalandBasicResearchonChronicDiseases,Tangshan063000,China.E-mail:ljkyxhljn@163.com)
AIM: To explore the role of imbalance of local renin-angiotensin system (RAS) in lung injury by observing the changes of angiotensin Ⅱ type 1 receptor (AT1R) and Mas receptor protein expression in the lung and the degree of lung injury subject to limb ischemia-reperfusion (LIR) in the mice. METHODS: Male ICR mice (n=42, 8 weeks old) were randomly assigned into 7 groups (6 in each group), including control group and 6 model groups with LIR of 0.5 h, 1 h, 2 h, 4 h, 6 h and 12 h reperfusion. Tourniquets were used to block the blood flow of the hind limbs of the ICR mice and were released after 2 h ischemia to initiate reperfusion. The mice were sacrificed by eyeball blood withdrawal at different time points after reperfusion. The organ coefficient and wet/dry weight ratio (W/D) of the lung tissue were calculated. Bronchoalveolar lavage fluid (BALF) was taken for cell counting and protein concentration measurement. The histopathological changes of the lung tissues was observed, and the pathological score was calculated. The protein expression of AT1R and Mas receptor in the lung tissues was determined by Western blot. RESULTS: The organ coefficient, W/D of lung tissue, and cell number and protein concentration in BALF of model groups were significantly higher than those in control group after LIR. The pathological changes were found in the lung tissue of model mice, including alveolar capillary dilation and congestion, edema, inflammatory cell infiltration in peripheral vascular, alveolar and bronchial walls, alveolar septal thickening and inflammatory cell infiltration. The lung injury score was elevated gradually along with the extension of reperfusion time. The protein expression of AT1R began to increase at reperfusion time points of 0.5 h and 1 h. With the extension of reperfusion time, the protein expression of AT1R decreased gradually. Conversely, the protein expression of Mas receptor increased gradually with prolonged reperfusion. CONCLUSION: LIR induces acute lung injury gradually. The imbalance of AT1R and Mas receptor expression may be involved in the damage process.
Ischemia-reperfusion; Lung injury; Renin-angiotensin system; AngiotensinⅡ type 1 receptor; Mas receptor
1000- 4718(2017)07- 1264- 07
2016- 09- 23
2017- 04- 05
国家自然科学基金资助项目(No. 81372029);河北省自然科学基金资助项目(No. H2015209153);国家级大学生创新创业训练计划项目(No. 201510081007); 河北省研究生创新资助项目(No. CXZZBS2017127)
R363
A
10.3969/j.issn.1000- 4718.2017.07.018
杂志网址: http://www.cjpp.net
△通讯作者 Tel: 0315-3725606; E-mail: ljkyxhljn@163.com
