琼胶降解菌FG12的全基因组测序
2017-07-18舒培军曾鸿俏张力雄刘明明方再光
舒培军,曾鸿俏,张力雄,刘明明,方再光
(海南大学 热带生物资源教育部重点实验室,海南省热带水生生物技术重点实验室,海南 海口570228)
琼胶降解菌FG12的全基因组测序
舒培军,曾鸿俏,张力雄,刘明明,方再光
(海南大学 热带生物资源教育部重点实验室,海南省热带水生生物技术重点实验室,海南 海口570228)
为了获得琼胶寡糖的高产琼胶降解菌,本研究通过Illumina Hiseq2000平台测序,使用SOAPdenovo 2.04软件(拼接组装),Glimmer 3.02预测基因的开放性阅读框,RNAmmer 1.2预测rRNA,tRNAscan-SE 1.23预测tRNA以及COG、CO和KEGG等来预测FG12的基因功能,并对其进行了全基因组测序,结果表明:FG12的基因组大小为4.11Mb,GC含量为37.76%;共有4 441个开放性阅读框,其平均长度为780 bp,有76个tRNA和5个rRNA;有与琼胶寡糖代谢相关的基因和抗生素的代谢途径,因此得出,琼胶降解菌FG12有改造成为高效工程菌的潜力.
琼胶降解菌; 琼胶寡糖; 全基因组测序; 基因功能
琼胶降解菌可产生琼胶酶,降解琼脂并生成琼胶寡糖.根据琼胶酶作用方式的不同,可将其分为α-琼胶酶和β-琼胶酶[1].α-琼胶酶可产生以3,6-内醚-α-L半乳糖为还原性末端的琼寡糖(agaro-oligosaccharides);β-琼胶酶则产生以β-D-半乳糖为还原性末端的新琼寡糖(neoagaro-oligosaccharades)[2].琼胶酶降解所产生的琼胶寡糖具有抗炎、抗病毒、抗氧化、增强免疫等作用[3],其在食品、药物及化妆品等行业具有非常好的应用前景,因此已经成为国内外研究的热点.但是,由于缺乏性状稳定且产酶能力强的琼胶降解菌,因此琼胶寡糖的应用受到了很大的限制.
全基因组De novo测序,也称为基因组从头测序,其先是在没有任何参照序列的情况下进行基因组测序,然后再利用生物信息学方面的技术拼接和组装已经获得的序列,进而获得其基因图谱[4].全基因组测序对全面了解一个物种的分子进化、基因组成和基因调控等有着非常重要的意义.随着基因组学和DNA测序技术的迅速发展,微生物的全基因组测序已经变得越来越便捷和高效.目前,已有类芽孢杆菌[6]、大丽轮枝菌[7]、多杀性巴氏杆菌[8]、鳜鱼弹状病毒[9]等微生物得到测序.尽管如此,然而关于热带琼胶降解菌全基因组测序的相关研究在国内仍尚未见报道.
为此,本研究通过全基因组测序,分析了高效琼胶降解菌Vibriosp.FG12[10]的基因组分,并通过与COG、CO和KEGG等数据库的比对预测了其基因功能,旨在通过基因改造来获得琼胶寡糖的高产菌株并为其提供理论依据.
1 材料与方法
1.1 菌株 以海南岛热带海洋环境分离和鉴定的琼胶降解菌Vibriosp.FG12为实验对象.
1.2 细菌基因组DNA的提取 待细菌达到纯培养后,挑单菌落进行液体培养,并在28 ℃,150 r·min-1摇床培养16 h后,按照细菌基因组DNA提取试剂盒的说明进行提取,同时将所提取的DNA用琼脂糖凝胶进行电泳检测.
1.3 全基因组测序 在所提取的基因组DNA经电泳检测后,对其DNA含量进行估计,并在其浓度达到深圳华大基因研究院所送样品的标准后,采用干冰保存并寄送至华大基因研究院进行全基因组测序.
1.3.1 序列组装 通过Illumina Hiseq2000平台测序,使用SOAPdenovo 2.04短序列组装软件进行组装.基于reads之间的overlap区,通过reads高通量测序平台产生的序列,拼接并获得Contigs,然后构建454 Paired-nd库以确定来自同一转录本的不同Contig的先后顺序,并利用先后顺序已知的Contigs来共同组成Scaffold.
1.3.2 基因预测 将Glimmer3.02预测基因ORFs,RNAmmer 1.2(http://www.cbs.dtu.dk/services/RNAmmer/)预测rRNA,tRNAscan-SE 1.23(http://gtrnadb.ucsc.edu/)预测tRNA与Rfam10.1数据库进行比对,寻找sRNA, TRF(Tandem Repeat Finder)4.04(http://rfam.sanger.ac.uk/),预测串联重复序列,并根据重复单元长度及数目筛选出其中的微卫星以及小卫星序列.
1.3.3 基因组功能注释 通过blastx将基因序列与NR数据库、京都基因和基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)、SwissProt、蛋白质直系同源基因簇(Cluster of Orthologous Groups of Proteins,COG)等进行比对,得到和给定序列具有最高序列相似性的蛋白,从而得到该基因的蛋白功能注释信息.根据NR的注释信息,通过预测得到基因的GO注释信息,再通过网络基因本体论注解绘图工具(Web Gene Ontology Annotation Plot,WEGO)对其进行详细的功能分类;再根据COG数据库的注释信息将所预测的功能分类;最后根据KEGG注释信息进一步得到基因的代谢途径.
2 结果与分析
2.1 基因组装结果的分析 基于软件SOAPdenovo 2.04的测序数据,组装并得到FG12基因组,大小为4.11Mb,GC含量为37.76%,共135个Scaffolds,299个Contigs(见表1).将所有拼接得到的Contigs按照从大到小进行排序,其累加片段的长度达到所有Contigs总长度的50%时,其所对应的Contig长度为N50值,它是评价序列组装好坏的一个指标.一般来说,N50越大,说明组装得越好,大片段的比例则越高[11].

表1 FG12组装结果
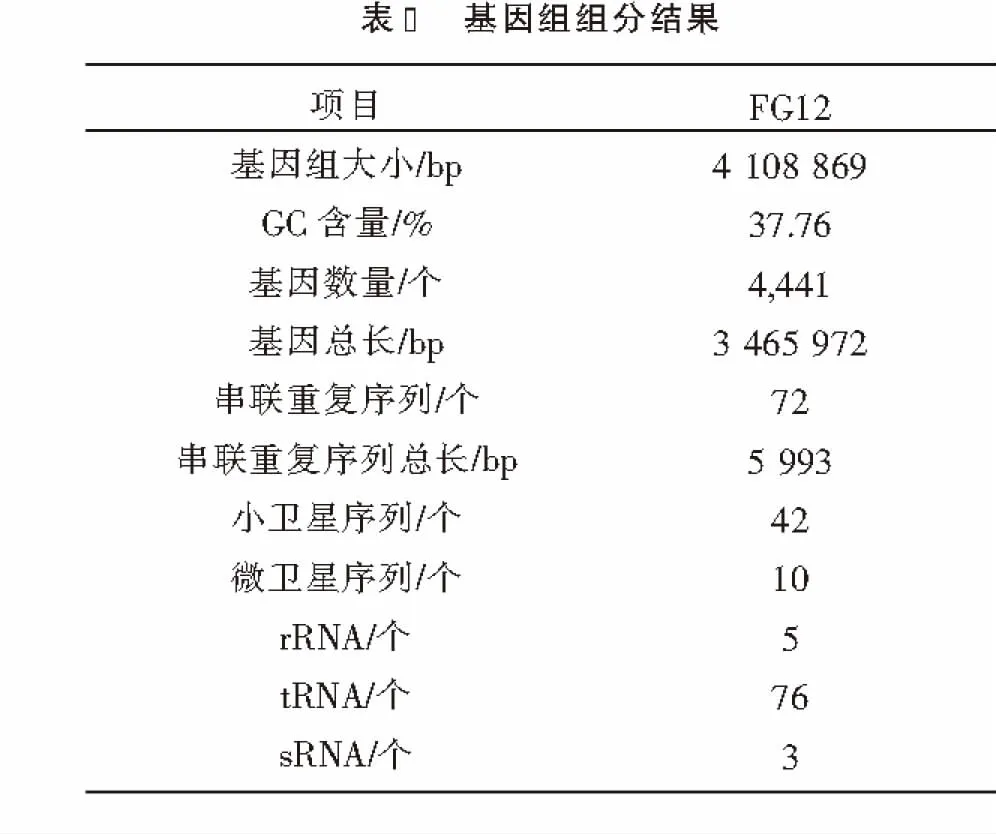
2.2 基因预测结果的分析 通过基因预测、重复序列预测、非编码RNA预测等方法来获取测序菌株基因组的组成情况(见表2).通过对基因组组分分析后发现,FG12的基因组含有4 441个基因,总长度为3 465 972 bp,平均长度780 bp,占基因组全长的84.35%;含有72个串联重复序列,总长为5 993bp,占基因组全长的0.145 9%;此外,还含有42个小卫星序列,10个微卫星序列以及76个tRNA和5个rRNA.
2.3 功能注释结果的分析 基因功能注释主要是通过与相应的蛋白质数据库进行比对来预测其可能的基因功能,然后对其功能进行分类和统计.
2.3.1 COG功能分类 由COG蛋白功能注释信息(见图1)可知:FG12最多的功能归类为通用功能(General Function Prediction Only),占12.85%;氨基酸转移与代谢(Amino Acid Transport And Metabolism)占10.16%;糖类的运输与代谢(Carbohydrate Transport And Metabolism)占6.68%;能量产生与转化(Energy Production And Conversion)占5.49%;信号转导机制(Signal Transduction Mechanisms)占3.81%.
2.3.2 GO功能注释 GO功能分类主要包括生物过程、细胞组分和分子功能三大类.GO功能分类图显示(见图2):细胞过程(Cellular Process)和代谢过程(Metabolic Process)在生物过程中十分活跃;细胞(Cell)和细胞部位(Cell Part)在细胞中占主导地位;结合(Binding)和催化活力(Catalytic Activity)在分子功能中起重要作用.
在FG12中,与生物过程(Biological Process)有关的基因有4 399条,与细胞组分(Cellular Component)有关的基因有1 951条,对应到分子功能(Molecular Function)的基因有3 085条.
2.3.3 KEGG代谢通路 根据KEGG的注释信息可知(见图3):参与膜运输(Membrane Transport)代谢途径的基因最多,其次为糖代谢(Carbohydrate Metabolism),再次为氨基酸代谢(Amino Acid Metabolism).FG12中参与糖代谢(Carbohydrate Metabolism)的基因有385个;参与氨基酸代谢(Amino Acid Metabolism)的基因有345个;参与酶家族(Enzyme Families)代谢的基因有96个.

代谢途径的分析表明:基因组含糖类代谢、能量代谢、脂类代谢、核苷酸和氨基酸代谢、次生代谢产物的合成代谢等基因.糖代谢途径中都包含了糖酵解途径、磷酸戊糖途径,此外还发现,FG12可以利用果糖、甘露糖、半乳糖、C5分支的二元酸等.在次生代谢产物的合成代谢途径中,FG12能合成头孢菌素、新生霉素、链霉素、丁苷菌素和新霉素.根据代谢途径的酶和基因的异同,图3用不同颜色来表示选中的代谢途径及酶的差异.
3 讨 论
利用全基因组测序技术对FG12基因组进行研究,可预测其基因组的组分,注释相应的功能基因,并探究其代谢途径.
FG12基因组大小为4.11 Mb,G+C含量为37.76%,共135个Scaffolds,299个Contigs,42个小卫星序列,10个微卫星序列,76个tRNA,5个rRNA.与嗜琼胶卵链菌(CatenovulumagarivoransYM01T)G+C的含量(为44.8%)[12]和白色噬琼胶菌(AgarivoransalbusMKT106)的G+C含量(为48%~50%)[13]相比,FG12的G+C含量偏低.
COG注释信息中,FG12含有21种COG功能类型,主要包括细胞代谢、细胞信号转导等.
`
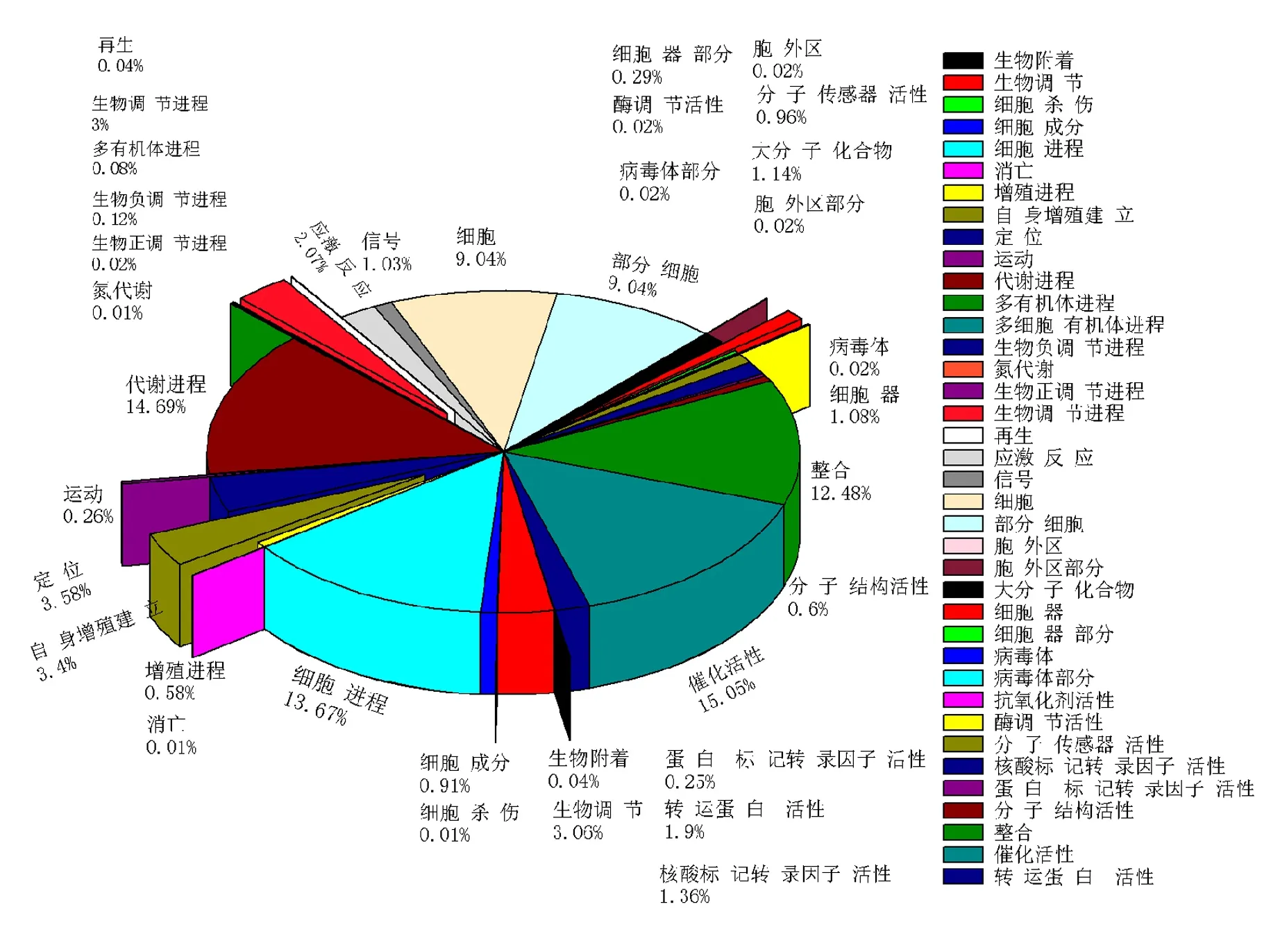
图2 FG12的GO功能注释

GO注释分别将FG12基因组注释到3大类39个COG功能亚类上, KEGG分析能把其定位到146个代谢通路中,包括物质代谢、次生代谢产物的生物合成等,其中参与糖代谢的基因为385条;参与氨基酸代谢的基因为345条.
目前,对于琼胶降解菌的全基因组测序尚无相关报道.高效海洋琼胶降解菌FG12的全基因组序列的测序将会为后续功能基因组学的研究提供理论依据,研究琼胶降解菌活性物质产生的调控机制将有助于海洋高效琼胶降解菌在工业生产的开发和应用.全基因组测序技术只是基因组学研究的起点,它所能解释的生物学现象和机制不多,在获得了细菌的基因组信息的基础上,如何去理解和消化乃至于运用庞大的信息依旧任重道远.
[1] Duckworth M, Turvey J R. The specificity of an agarase from aCytophagaspecies[J]. Biochemical Journal, 1969, 113(4): 693-696.
[2] Arnott S, Fulmer A, Seott W E, et al. The agarose double helix and its function in agarose gel structure[J]. Journal of Molecular Biology, 1974, 90(2): 269-284.
[3] Wang J, Ma J F, Miao T T, et al. Cloning and expression ofAgarivoransalbusQM38β-agarase gene agaDO2[J]. Marine Sciences, 2010(8):6-10.
[4] 聂小军. 基于高通量测序技术的小麦和紫茎泽兰基因组学初步研究[D]. 西安:西北农林科技大学, 2013.
[5] 刘蓉蓉. 高等植物基因组测序回顾与展望[J]. 生物技术通报, 2011(5): 10-14.
[6] 夏天.Paenibacillussp.Aloe-11全基因组测序及比较基因组学研究[D]. 重庆:西南大学,2012.
[7] 陈相永. 大丽轮枝菌不同毒力菌株全基因组测序及重测序分析[D].北京:中国农业科学院,2012.
[8] 刘文静. 多杀性巴氏杆菌全基因组测序与比较基因组学分析[D]. 武汉:华中农业大学, 2012.
[9] 陶建军. 鳜鱼弹状病毒的分离、鉴定及基因组测序[D]. 北京:中国科学院水生生物研究所,2006.
[10] 尹群健,陈潇骁,杨宏胜,等. 琼胶降解菌产酶条件优化、酶学性质和糖代谢途径[J]. 广东农业科学,2014(9):117-122.
[11] 黄勇. 基于高通量测序的微生物基因组学研究[D]. 北京:军事医学科学院, 2013.
[12] 严书林. 3株新菌的分类鉴定及新型耐热琼胶酶基因的克隆表达[D].青岛:中国海洋大学, 2011.
[13] Midori K, Akira Y.Agarivoransalbusgen. nov. sp. nov. aγ-proteobacterium isolated from marine animals[J]. International journal of systematic and evolutionary microbiology, 2004, 54: 693-697.
Whole Genome Sequencing of Agar-degrading Strain FG12
Shu Peijun, Zeng Hongqiao, Zhang Lixiong, Liu Mingming, Fang Zaiguang
(Key Laboratory of Tropical Biological Resources of Ministry of Education, Key Laboratory of Biotechnology of Tropical Aquatic Organisms of Hainan Province, Hainan University, Haikou 570228, China)
To obtain agar-degrading strain high-producing agarooligosaccharide, the genome of FG12 was sequenced on the Illumina Hiseq2000 sequencing platform. SOAP denovo 2.04 was used for assembling. Glimmer 3.02 was used to predict open reading frame of the gene. RNAmmer 1.2 was used to predict rRNA. tRNAscan-SE 1.23 was used to predict tRNA. Gene function was annotated by COG, CO, and KEGG databases. The results showed that the genome size of FG12 was 4.11 Mb with the G+C content of 37.76%, containing 4,441 open reading frames (ORFs), the average length of which was 780 bp, and which included 76 tRNA and 5 rRNA. Agar oligosaccharide metabolism-related genes and metabolic pathways of antibiotics were observed. These data suggested that Agar-degrading bacteria FG12 has the potential to be transformed into efficient engineering bacteria.
agar-degrading strain; agarooligosaccharide; whole genome sequencing; gene function
2017-02-27
海南大学青年基金(qnjj1205),校县合作项目(02005001)
舒培军(1988-),男,贵州兴义人,海南大学海洋学院2012级硕士研究生,E-mail: xiubinsharp@126.com
方再光(1975-),男,湖南新化人,副教授,研究方向:海洋生物技术,E-mail: guangyan0508@163.com
1004-1729(2017)02-0140-05
Q523+.8
A DOl:10.15886/j.cnki.hdxbzkb.2017.0025
