伊潘立酮-羟丙基- β-环糊精包合物的制备及表征、肌肉刺激和药代动力学研究
2017-07-12欧云国何海云单雪峰黄华
欧云国,何海云,单雪峰,黄华
(1.重庆医药工业研究院有限责任公司,重庆400061;2.重庆医科大学,重庆400016)
·实验研究·
伊潘立酮-羟丙基- β-环糊精包合物的制备及表征、肌肉刺激和药代动力学研究
欧云国1,何海云2,单雪峰2,黄华2
(1.重庆医药工业研究院有限责任公司,重庆400061;2.重庆医科大学,重庆400016)
目的通过对伊潘立酮与羟丙基- -环糊精(HP- -CD)包合作用进行对比研究,达到制备安全、有效的肌肉注射给药制剂的目的。方法分别研究伊潘立酮在HP- -CD水溶液与HP- -CD 0.2%磷酸溶液中的相溶解度曲线;采用溶液搅拌法,冷冻干燥技术制备包合物;采用红外、分光光度法、差示扫描量热分析法(DSC)、溶解度法对包合物进行验证;肌肉注射给予伊潘立酮-HP- -CD包合物溶液,对兔股四头肌进行肌肉刺激性试验;对比格犬进行伊潘立酮-HP- -CD包合物溶液肌肉注射给药和伊潘立酮片口服给药,对药代动力学指标进行研究。结果相溶解度曲线呈Ap型,表明客分子与主分子包合比例为1∶n(n 2);红外、DSC、溶解度试验结果表明,伊潘立酮包合于HP- -CD的空腔结构中;包合物溶液对肌肉注射部位无刺激性;药代动力学结果表明,伊潘立酮与HP- -CD包合后,肌肉注射给药药物吸收较快,与口服片剂相比,其相对生物利用度为143%。结论伊潘立酮-HP- -CD包合物可显著提高溶解度,能很好地应用于注射给药途径。
羟丙基- -环糊精;伊潘立酮;包合物;注射剂
临床常用的抗精神病药分为典型和非典型两类。非典型抗精神病药治疗谱广,对阴性症状效果优于典型药物,且安全性高,不良反应轻微,服用剂量小,锥体外系不良反应少,已成为临床一线用药[1]。伊潘立酮为新开发的非典型抗精神病药物,2009年美国食品药物管理局(FDA)批准伊潘立酮片上市,目前上市的制剂中没有其他给药途径[2]。伊潘立酮为混合型多巴胺D2及5-羟色胺(5-HT)2A型受体阻断剂,是一种白色至淡白色细结晶粉末,难溶于水,在0.1mol/L HCl中微溶,易溶于氯仿、乙醇、甲醇和乙腈。伊潘立酮片胃肠道吸收较好,血浆达峰时间为2~4 h,3~4 h内达到稳态浓度。但因其首过效应,伊潘立酮的口服生物利用度低,小鼠<1%,大鼠为5%,兔和犬为19%,人约为36%[3]。据文献报道,伊潘立酮治疗精神分裂症效果好,毒副作用低[2-4],尤其对急性激越症状的精神分裂症患者有效[5]。制备伊潘立酮的包合物,增加其溶解度,使其能应用于伊潘立酮注射剂的制备,以达到起效快、提高生物利用度、减少给药剂量的目的,对增加伊潘立酮在临床的应用具有重要意义。
目前,环糊精包合技术已得到了广泛应用。环糊精是内径为0.6~1.0 nm的环状中空的圆筒状分子结构,其内疏水其外亲水。适合形状和大小的亲脂性分子或基团进入环糊精的亲脂性空腔后产生包合作用[6],难溶性药物包合后,可使药物的溶解度增加,生物利用度提高[7]。毒理学研究表明,天然的β-环糊精不能应用于肠道外给药(注射给药),而其亲水性衍生物HP-β-CD具有更强的包合能力,更大的溶解度,且采用非肠道给药的低毒性对人无不良反应[8]。且因HP-β-CD有适合的空腔大小和低的价格,它已成为CD衍生物中最常用的包合材料,许多上市制剂已用HP-β-CD制备注射剂[9-10],如国外上市制剂伊曲康唑包合物注射液(商品名Sporanox)。因此,本研究以HP-β-CD为包合材料,用于伊潘立酮包合物注射剂的研究。
笔者采用溶液搅拌法,制备伊潘立酮与HP-β-CD包合物溶液(规格6 g/L),并采用冷冻干燥技术,制备得包合物固体粉末;相溶解度法研究其包合作用;采用红外分光光度法、差示扫描量热分析法(DSC)、溶解度法对包合物进行验证;并研究包合物溶液(6 g/L)对注射部位(肌肉)的刺激性,以及采用高效液相色谱法对比格犬进行伊潘立酮-HP-β-CD包合物溶液肌肉注射给药和伊潘立酮片口服给药的药代动力学研究。现报道如下。
1 仪器与试药
UV-2015PC型紫外分光光度计(日本岛津公司);DF-101S型集热式恒温加热磁力搅拌器(郑州长城科工贸有限公司);ALPHA 1-2LD型冷冻干燥机(德国Christ公司);STA-449C型差示扫描量热仪(德国Netzsch公司);AVATAR 330FT-IR型红外光谱仪;A200S型分析天平(德国Sartorius公司);HZ-881S台式水浴恒温振荡器(太仓市科学实验仪器厂)。伊潘立酮(上海诺特生物科技有限公司,批号为20110225,含量>99%);HP-β-CD(ISP国际特品公司);所用试剂均为分析纯。
2 方法与结果
2.1 标准曲线的建立
采用紫外分光光度法进行相溶解度研究,建立标准曲线。精密称取伊潘立酮对照品适量,用无水乙醇溶解并稀释成0.5 g/L的贮备液。分别精密移取贮备液0.1,0.2,0.3,0.4,0.5,0.6m L,置10 mL容量瓶中,按以下方法绘制2条标准曲线。标准曲线1∶80%乙醇稀释至刻度,照紫外分光光度法,在275 nm波长处测定吸光度;标准曲线2:无水乙醇稀释至刻度,同法在275 nm波长处测定吸光度。分别以吸光度(Y)为纵坐标、质量浓度(X)为横坐标进行线性回归,得标准曲线1方程Y=0.039X+0.002 7,r=0.998 6(n=6)和标准曲线2方程Y=0.039 1X-0.003 9,r=0.999 9(n=6)。结果表明,曲线1与曲线2质量浓度在25~30μg/mL范围内与伊潘立酮吸光度线性关系良好。
2.2 包合溶剂的选择
由于伊潘立酮在水中溶解度低,若以HP-β-CD水溶液进行包合,以分子状态存在的伊潘立酮较少,不利于包合反应的进行。因此应选择适当的溶剂,使伊潘立酮较好地溶解,以达到较好的包合效果。
分别以pH=2.0的枸橼酸溶液、pH=2.0的酒石酸溶液、pH=2.0的盐酸溶液、pH=2.0的磷酸溶液、pH=2.0的甲磺酸溶液和水为溶剂,配制4.5%的HP-β-CD溶液,精密称取过量的伊潘立酮对照品(30 mg)于5m L上述溶液中,37℃恒温搅拌2 h后,过滤,取续滤液,无水乙醇稀释,照紫外分光光度法,275 nm波长处测定吸光度。根据标准曲线2计算溶解度,测得伊潘立酮溶解度(见表1)。结果表明,以HP-β-CD磷酸溶液为溶剂,伊潘立酮溶解度最大,因此选择pH=2.0的磷酸溶液(0.2%)为溶剂。

表1 不同溶剂的伊潘立酮溶解度
2.3 相溶解度曲线
照Higuchi和Connors的相溶解度试验方法进行相溶解度试验[11]。将过量的伊潘立酮对照品(30 mg)加入5 m L一系列不同浓度的HP-β-CD水溶液中(0~4.0%)。分别于25,37,60℃恒温振荡器振荡7 d后,0.45μm微孔滤膜过滤,取续滤液2 mL于10 m L容量瓶中,无水乙醇稀释至刻度,照紫外分光光度法,在275nm波长处测定吸光度,根据标准曲线1计算药物溶解度。
照同样方法,在0.2%磷酸水溶液中进行相溶解度试验。分别在45,60℃恒温条件下,将过量的伊潘立酮对照品(30 mg)加入5 m L一系列不同体积分数(0~4.0%)的HP-β-CD 0.2%磷酸溶液中。分别于25,37,60℃恒温振荡器振荡7 d后,0.4%氢氧化钠溶液调节pH为5.5,0.45μm微孔滤膜滤过,取续滤液0.1mL置20 mL容量瓶中,无水乙醇稀释至刻度,照紫外分光光度法,在275 nm波长处测定吸光度,根据标准曲线2,计算药物溶解度。
以HP-β-CD浓度(X,%)为横坐标、伊潘立酮质量浓度(Y,μg/mL)为纵坐标绘制相溶解度曲线,并对曲线中成线性关系的点进行线性回归,采用以下公式计算各个曲线的表观稳定常数。
stability constant(Kc)=[slope/So(1-slope)]
其中,So为相应条件下伊潘立酮的饱和溶解度,slope为曲线的斜率。
根据不同温度下包合物的表观稳定常数Kc,由公式ΔG=-RTlnKc计算不同温度下包合过程的吉布斯自由能变化ΔG。再根据Van′tHoff方程lnKc=-ΔH/RT+ΔS/R,以lnKc对1/T做线性回归,由回归方程的斜率(ΔH/R)与截距(ΔS/R)即可求得包合过程中的焓变(ΔH)与熵变(ΔS)。分别为以水为溶剂和以0.2%磷酸为溶剂的伊潘立酮-HP-β-CD相溶解度图见图1和图2。
对比以水为溶剂和以0.2%磷酸为溶剂相溶解度曲线,可知相同浓度的HP-β-CD溶液,以0.2%磷酸为溶剂时伊潘立酮的溶解度远远大于以水为溶剂时伊潘立酮的溶解度。由图1可知,添加过量伊潘立酮,随着HP-β-CD浓度的增加,伊潘立酮的溶解度增大速度加快。由图2可见,固定伊潘立酮的量,随着HP-β-CD浓度的增加,伊潘立酮的溶解度增大速度加快,直到伊潘立酮与HP-β-CD分子比为1∶6时,伊潘立酮溶解完全,浓度不再增加,因此可知此比例下伊潘立酮溶解度最大。根据Higuchi和Connors的理论,以水为溶剂或以0.2%磷酸为溶剂,相溶解度图均为Ap型,表明客分子与主分子包合比例为1∶n(n≥2)。以水为溶剂,对比25,37,60℃曲线,温度增高,HP-β-CD的增溶能力上升,即增高温度有利于药物的包合。以0.2%磷酸为溶剂,对比45℃与60℃曲线,相同浓度下45℃伊潘立酮溶解度更大。分别计算各个条件下的稳定常数和热力学参数见表2。可见,ΔH>0,说明反应为吸热反应;ΔG<0,说明本试验条件下,反应为自发进行;另ΔG随温度升高负值增大,说明温度升高,形成包合物的自发倾向大。

图1 以水为溶剂的伊潘立酮相溶解度图

图2 以0.2%磷酸为溶剂的伊潘立酮相溶解度图
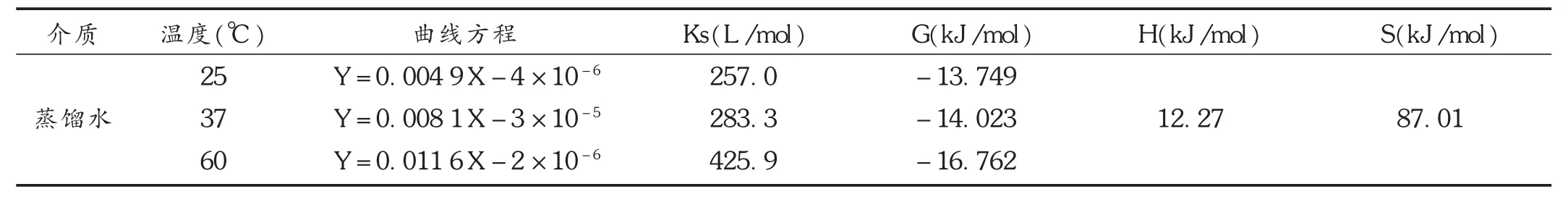
表2 热力学参数
2.4 伊潘立酮-HP- -CD包合物的制备
根据相溶解度试验结果,确定伊潘立酮与HP-β-CD的分子比例为1∶6。精密称取HP-β-CD适量,置50 mL烧杯中,加0.2%磷酸水溶液10m L,搅拌溶解后,加入伊潘立酮60mg(即6 g/L),置恒温磁力搅拌器上,250 r/min,50℃搅拌30min,0.4%氢氧化钠溶液调节pH为5.5,0.22μm微孔滤膜滤过,即得包合物溶液,取滤液冷冻干燥,即得包合物固体粉末。
2.5 伊潘立酮HP- -CD物理混合物的制备
按照1∶6(客分子∶主分子)的分子比例,分别精密称取HP-β-CD与伊潘立酮适量,置研钵中,研磨混合10 min,得均匀的伊潘立酮HP-β-CD物理混合物。
2.6 差示扫描量热分析
采用STA-449C型差示扫描量热仪(德国Netzsch公司),对伊潘立酮、HP-β-CD、伊潘立酮与HP-β-CD物理混合物、伊潘立酮与HP-β-CD包合物进行差示扫描量热分析(DSC)。升温速率为10℃/min,升温范围为0~450℃,测定气体为氮气(20m L/min)。
通过DSC法可以说明伊潘立酮与HP-β-CD的包和作用。当客分子包合于主分子的空腔结构中后,客分子的熔点、沸点、升华点会发生转移或消失。伊潘立酮、HP-β-CD、物理混合物、包合物的DSC图谱见图3。可见,伊潘立酮在130℃与250℃附近分别为吸热峰与放热峰;HP-β-CD在50℃与350℃附近有特征吸热峰;混合物在50,350℃有吸热峰,且与伊潘立酮一样,在130℃也有吸热峰,说明物理混合物为伊潘立酮与HP-β-CD特征峰的叠加;包合物与物理混合物相比,包合物图谱中药物130℃的吸热峰消失,350℃处放热峰移至270℃,说明伊潘立酮与HP-β-CD存在相互作用,并且可能是由于伊潘立酮已经包合于HP-β-CD的空腔结构中,伊潘立酮吸热峰消失。

1.HP- -CD 2.伊潘立酮3.物理混合物4.包合物图3差示扫描量热图
2.7 红外分光光度法
采用AVATAR 330FT-IR型红外光谱仪,用KBr分别将伊潘立酮、HP-β-CD、伊潘立酮和HP-β-CD物理混合物、伊潘立酮和HP-β-CD包合物研磨压片。在4 000~400 cm-1光谱范围内进行红外光谱扫描,记录红外吸收图谱。伊潘立酮、HP-β-CD、物理混合物、包合物的红外图谱见图4。可见,HP-β-CD在3 475,2 925 cm-1处可见游离-氢氧基和缔合-氢氧基的振动吸收峰,伊潘立酮在3030 cm-1,1 680~1 500 cm-1,和900~690 cm-1处分别有Ar—H键的伸缩振动,芳烃C=C键的伸缩振动和Ar—H键的面外弯曲振动吸收峰。物理混合物图谱为伊潘立酮与HP-β-CD特征吸收峰的叠加,如1 510,1 151,1 084,1 032 cm-1。包合物图谱中伊潘立酮的特征吸收峰消失,图谱与HP-β-CD基本相同,说明伊潘立酮与HP-β-CD产生了包合作用。
2.8 溶解度测定
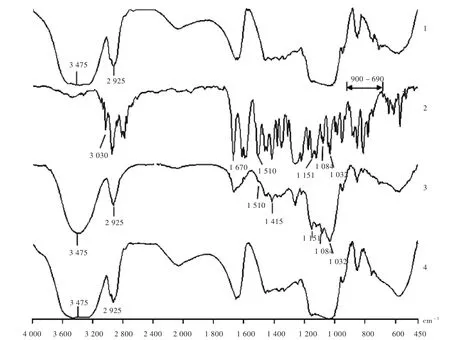
1.HP- -CD 2.伊潘立酮3.物理混合物4.包合物图4红外扫描图谱
分别取过量伊潘立酮、伊潘立酮与HP-β-CD物理混合物、伊潘立酮与HP-β-CD包合物,置试管中,加水配制成过饱和溶液,25℃水浴静置12 h,过滤,取续滤液,无水乙醇稀释成适量浓度,275 nm波长处测定吸光度,计算溶解度。计算得伊潘立酮、伊潘立酮与HP-β-CD物理混合物、伊潘立酮包合物的溶解度分别为10,146,19 500μg/mL,可见混合物的的溶解度为原来的14.6倍,而包合物溶解度为原来的1 950倍,包合后伊潘立酮的溶解度大大增加。
2.9 兔肌肉刺激性试验
药物的水溶性是影响肌肉注射给药生物利用度的关键。药物包合后,在生理的pH组织间液中,包合的药物可能从包合材料游离出来而析出沉淀,造成注射部位的刺激性以及影响药物的扩散和吸收。因此进行肌肉刺激性试验,考察制备得的伊潘立酮包合物是否适合于肌肉注射。
取家兔4只,每只家兔左侧股四头肌注射生理盐水,右侧股四头肌注射伊潘立酮-HP-β-CD包合物溶液(6 g/L),分别于第1,2,3,4天注射0.2,0.4,0.8,1.0mL(避免直立性低血压)。注射后观察注射部位有无充血、水肿等反应。最后1次给药48 h后处死半数动物,纵向切开皮肤肉眼观察两侧注射部位,看有无充血、水肿,并取其组织做病理学检查。按以下标准评价该药的刺激反应:0级,给药部位无明显反应;1级,给药部位轻度充血,水肿直径<0.15 cm;2级,给药部位中度充血,水肿直径0.5~1.0 cm;3级,给药部位重度充血、红肿,肌肉有变性;4级,出现肌肉褐色变性、坏死;5级,肌肉严重变性,出现大片坏死。剩下半数动物继续观察14 d后重复上述检查。
给药后剪取左右两侧股四头肌肉眼观察,未见明显刺激反应,反应分级结果见表3。反应级数之和为1,小于10,说明没有刺激性。1号与2号家兔股四头肌的显微镜镜检图分别见图5和图6。可见,试验侧与对照侧比较未见明显病理学变化,镜检肌纤维组织横纹清晰、结构完整,无变性、坏死和炎性反应。
表3 兔肌肉刺激性反应分级结果
2.10 犬体内药代动力学研究
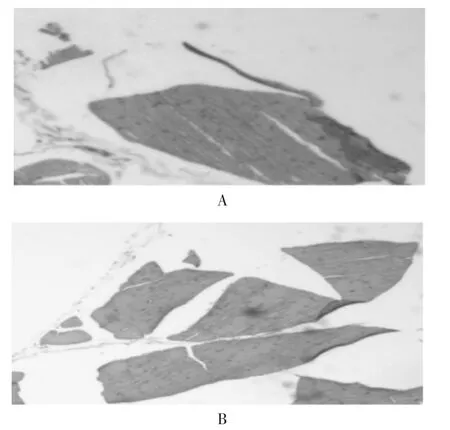
A.1号家兔B.2号家兔图5左侧股四头肌镜检图

A.1号家兔B.2号家兔图6右侧股四头肌镜检图
取规格为6 g/L的伊潘立酮包合物溶液与规格为8 mg/片的伊潘立酮片进行试验,给药剂量为8 mg。试验在重庆医药工业研究院进行。取平均体质量为10~12 kg的比格犬6只。随机分为两组,采用双周期两制剂交叉试验设计,洗净期为1周。试验前所有动物禁食1晚,但均可自由饮水。分别对两组动物进行肌肉注射伊潘立酮-HP-β-CD包合物溶液1.35m L(8mg)和口服伊潘立酮片1片(8 mg/片)。给药后分别于0,0.83,0.25,0.5,0.75,1,1.5,2,3,4,6,8,12,24 h时腿静脉取血3mL,置肝素真空采血管中,4 000 r/min离心10min,取上层血浆备用。
采用高效液相色谱(HPLC)法测定血浆中伊潘立酮的药物质量浓度。仪器为日本岛津高效液相色谱仪(LC-10泵,SPD-10A型可调波长紫外检测器)。依利特色谱C18柱(250mm×4.6mm,5μm);流动相为乙腈-0.1 mol/L NaH2PO4(磷酸调pH值为3.0)-甲醇(34∶60∶10);流速1 m L/mim;检测波长230 nm;进样量100μL。血浆样品测定方法:取血浆1m L,加10μg/mL的齐拉西酮内标溶液50μL,混匀,加二氯甲烷4m L,旋转振荡2min,3000 r/min离心5min,取有机层置40℃水浴中氮气吹干,150μL流动相复溶,取100μL注入液相色谱仪。结果表明,伊潘立酮在25~350 ng/m L质量浓度范围内呈良好线性关系(Y=240.5X-6.813 3,R2=0.998 2),平均回收率为90.87%,精密度的RSD为7.36%,LLOQ为10 ng/m L。
伊潘立酮-HP-β-CD包合物溶液(1∶6,6mg/mL)与伊潘立酮片(8 mg/片)相同剂量给药后,比格犬体内的平均血浆药物质量浓度-时间曲线见图7,药代动力学参数见表4。可见,肌肉注射伊潘立酮-HP-β-CD包合物,其血药峰浓度(Cmax)为口服片剂的3.29倍。注射给予伊潘立酮包合物,其0-∞药时曲线下面积(AUC0-∞)为口服片剂的1.43倍,生物利用度增加。
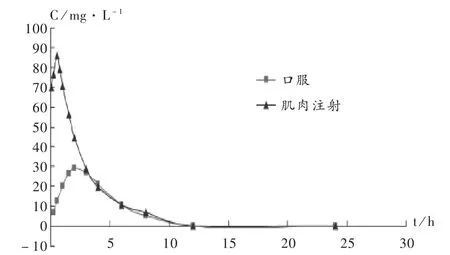
图7 平均血浆药物质量浓度-时间曲线

表4 药代动力学参数
肌肉注射给药,药物的溶解和扩散是药物吸收的一个重要因素,由表4可见,伊潘立酮-HP-β-CD包合物注射给药后,能在注射部位很好地吸收,达到了起效快和提高生物利用度的目的。根据结果可以推测,伊潘立酮与HP-β-CD产生包合作用,药物溶解度增加,在注射部位能较好地溶解和扩散;伊潘立酮在注射部位的生理pH条件下没有析出沉淀,吸收较快。
3 讨论
本研究结果显示,相溶解度图为Ap型。以0.2%磷酸水溶液为溶剂,分子比为1∶6(客分子∶主分子),采用溶液搅拌法可成功地制备得伊潘立酮-HP-β-CD包合物,并通过DSC、红外分光光度法进行了验证;溶解度测定结果表明,包合后溶解度增加了1 950倍。包合物溶液肌肉注射给药,对肌肉无刺激性。在比格犬体内的药代动力学研究表明,肌肉注射给药与口服给药相比,起效快,Cmax和生物利用度增加。
因此,通过制备伊潘立酮-HP-β-CD包合物,提高溶解度,使注射给药途径成为可能,拓宽了本品的临床应用途径,具有重要的临床意义和商业价值。
[1]Davis KL,Charney D,Coyle JT,et al.Neuropsychopharmacology:The Fifth Generation of Progress[M].Pennsylvania:Lippincott W illiams&W ilkins,2002:775-808.
[2]USFood&DrugAdministration.Lable[EB/OL].[2009-05-06].https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/0221921bl.pdf.
[3]US Food&Drug Administration.Pharmacology Review[EB/OL].[2009-05-06].https://www.accessdata.fda.gov/drugsatfda_ docs/nda/2009/022192s000_PharmR_P1.pdf,https://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/022192 s000_ PharmR_P2.pdf,https://www.accessdata.fda.gov/drugsatfda_ docs/nda/2009/022192s000_Pharm R_P3.pdf.
[4]Kane JM,Lauriello J,Laska E,et al.Long-term efficacy and safety of iloperidone:results from 3 clinical trials for the treatment ofschizophrenia[J].JClin Psychopharmacol,2008,28:S29-S35.
[5]Fanapt.For acute treatment of schizophrenia in adults[EB/OL].[2010-2-6](2011-5-3).http://www.fd.goy.
[6]Loftsson T,Brewster ME.Pharmaceuticalapplications of cyclodextrins.1.Drug solubilization and stabilization[J].J Pharm Sci,1996,85:1017-1025.
[7]SzejtliJ.Introductionandgeneraloverview cyclodextrinchemistry[J].Chem Rev,1998,98:1743-1753.
[8]Brewster M,Loftsson T.Cyclodextrins as pharmaceutical solubilizers[J].Adv Drug Del Rev,2007,59:645-666.
[9]Loftsson T,Duchene D.Cyclodextrins and their pharmaceutical applications[J].Int JPharm,2007,329:1-11.
[10]Gould S,Scott RC.2-Hydroxypropyl- -cyclodextrin(HP--CD):a toxicology review[J].Food Chem Toxicol,2005,43:1451-1459.
[11]Higuchi T,Connors K.Phase-solubility techniques[J].Adv nal Chem Instr,1965,4:117-212.
Preparation,Characterization,M uscular Stim ulation and Pharm acokinetics of the Inclusion Com p lex of Iloperidone-Hydroxyp ropyl-β-Cyclodextrin
Ou Yunguo1,He Haiyun2,Shan Xuefeng2,Huang Hua2
(1.Chongqing Pharmaceutical Research Institute Co.,Ltd.,Chongqing,China 400061;2.Chongqing Medical University,Chongqing,China 400016)
Objective To study the inclusion complex of Iloperdone-Hydroxypropyl-β-Cyclodextrin(HP-β-CD),in order to prepare a stable and effective parenteral formulation.M ethods Phase solubility study was carried out using distilled water and 0.2%phosphoric acid solution as dissolution medium,respecticely;the solution method and lyophilization techniques were used for inclusion complex preparation.The complexes were characterized by fourier transform infrared spectroscopy and differential scanning calorimetry studies and the solubility was determined by UV spectroscopy.The muscular stimulation test of Iloperdone-HP-β-CD inclusion complex solution was perform in rabbits,the pharmacokinetic of Iloperdone tablet and Iloperdone inclusion complex were evaluated after oral administration and muscular injection in dogs.Resu lts The phase-solubility profiles were classified as Ap type,indicating the formation of 1∶n(n≥2)stoichiometric inclusion complexes.The complexes were characterized by fourier transform infrared spectroscopy(IR)and differential scanning calorimetry(DSC)indicated that Iloperdone was able to form an inclusion complex with HP-β-CD.Iloperdone-HP-β-CD inclusion complex solution had no significant stimulation on muscles.In vivo pharmacokinetic study showed that the absorp tion rates of Iloperdone across muscles tissues were enhanced compared with oral administra tion.The relative bioavailabity was 143%.Conclusion The Iloperdone-HP-β-CD complex can significantly improve the sotubility,it is an attractive formulation for use in the parenteral.
HP-β-CD;Iloperdone;inclusion complex;injection
R943;R969.1
A
1006-4931(2017)10-0021-06
2017-01-29)
10.3969/j.issn.1006-4931.2017.10.006
欧云国,男,硕士研究生,工程师,研究方向为药物新制剂与新剂型,(电子信箱)ouyunguo@cpri.com.cn。
