RP-HPLC法测定洛索洛芬钠片的含量及有关物质
2017-07-07袁浩宇核工业四一六医院成都610051
袁浩宇,王 鹏,易 红,俞 瑜(核工业四一六医院,成都 610051)
RP-HPLC法测定洛索洛芬钠片的含量及有关物质
袁浩宇*,王 鹏,易 红#,俞 瑜(核工业四一六医院,成都 610051)
目的:改进测定洛索洛芬钠片中主成分含量及有关物质的方法。方法:采用反相高效液相色谱法。色谱柱为迪马Inspire C18,流动相为乙腈-0.01mol/L磷酸二氢钾溶液(含0.2%三乙胺,磷酸调节pH至3.0±0.1)(62∶38,V/V),流速为1.0m L/ min,柱温为40℃,检测波长为221 nm,进样量为20μL。结果:洛索洛芬钠峰与相邻杂质峰达到良好分离(分离度>1.5);洛索洛芬钠检测质量浓度线性范围为30.0~90.0μg/m L(r=0.999 8);洛索洛芬钠检测限为0.3μg/m L;精密度、稳定性、重复性试验的RSD<1.0%;回收率为99.00%~99.87%,RSD=0.33%(n=9)。结论:该方法准确、简便、快速,适用于洛索洛芬钠片的质量控制。
反相高效液相色谱法;洛索洛芬钠片;含量;有关物质
洛索洛芬钠(Loxoprofen sodium),化学名为2-[4-(2-氧代环戊烷-1-基甲基)苯基]丙酸钠二水合物,为前列腺素(PG)合成酶抑制剂,具有解热、镇痛及抗炎作用,临床上广泛用于类风湿性关节炎,腰痛,肩周炎,颈肩腕综合征,急性上呼吸道炎症,手术、外伤后及拔牙后的镇痛消炎,与同类药物相比具有起效更快、临床效果更好、副作用更小等特点[1]。为了保证用药安全、有效,笔者改进了以反相高效液相色谱法(RP-HPLC)测定洛索洛芬钠片中主成分含量及有关物质的方法,现报道如下。
1 材料
1.1 仪器
1100型HPLC仪,包括紫外检测器及ChemStation色谱工作站等(美国Agilent公司);FE20K型酸度计(瑞士Metter-Toledo公司);BS224S型万分之一电子天平(德国Sartorius公司)。
1.2 药品与试剂
洛索洛芬钠对照品(中国食品药品检定研究院,批号:100638-200407,纯度:99.0%,供含量测定用);洛索洛芬钠片(四川省人民医院制剂室自制,批号:110110、110111、110112,规格:60mg);乙腈(色谱纯,美国Fisher公司);其他试剂均为分析纯,水为自制超纯水。
2 方法与结果
2.1 色谱条件
色谱柱:迪马Inspire C18(250mm×4.6mm,5μm);流动相:乙腈-0.01mol/L磷酸二氢钾溶液(含0.2%三乙胺,磷酸调节pH至3.0±0.1)(62∶38,V/V);流速:1.0m L/min;检测波长:221 nm;柱温:40℃;进样量:20 μL。
2.2 溶液的制备
2.2.1 供试品溶液 取样品10片(批号:110110),精密称定,研细,精密称取细粉适量(约相当于洛索洛芬钠30mg),置于50m L量瓶中,加流动相溶解并稀释至刻度,摇匀,滤过,精密量取续滤液5m L,置于50m L量瓶中,加流动相稀释至刻度,摇匀,作为供试品溶液。
2.2.2 对照品溶液 精密称取洛索洛芬钠对照品15 mg,置于25m L量瓶中,加流动相溶解并稀释至刻度,摇匀,精密量取5m L,置于50m L量瓶中,加流动相稀释至刻度,摇匀,制成每1m L中含洛索洛芬钠60.0μg的溶液,作为对照品溶液。
2.2.3 空白辅料溶液 按样品处方精密称取除洛索洛芬钠外的空白辅料适量,再按“2.2.1”项下方法进行制备,即得空白辅料溶液。
2.3 系统适用性与专属性试验
2.3.1 系统适用性试验 精密量取“2.2”项下供试品溶液、对照品溶液和空白辅料溶液各20μL,分别按“2.1”项下色谱条件进样测定,记录色谱,详见图1。结果表明,理论板数按照洛索洛芬钠峰计算≥4 000,洛索洛芬钠峰的保留时间约为8.1min,与相邻杂质峰的分离度>1.5。
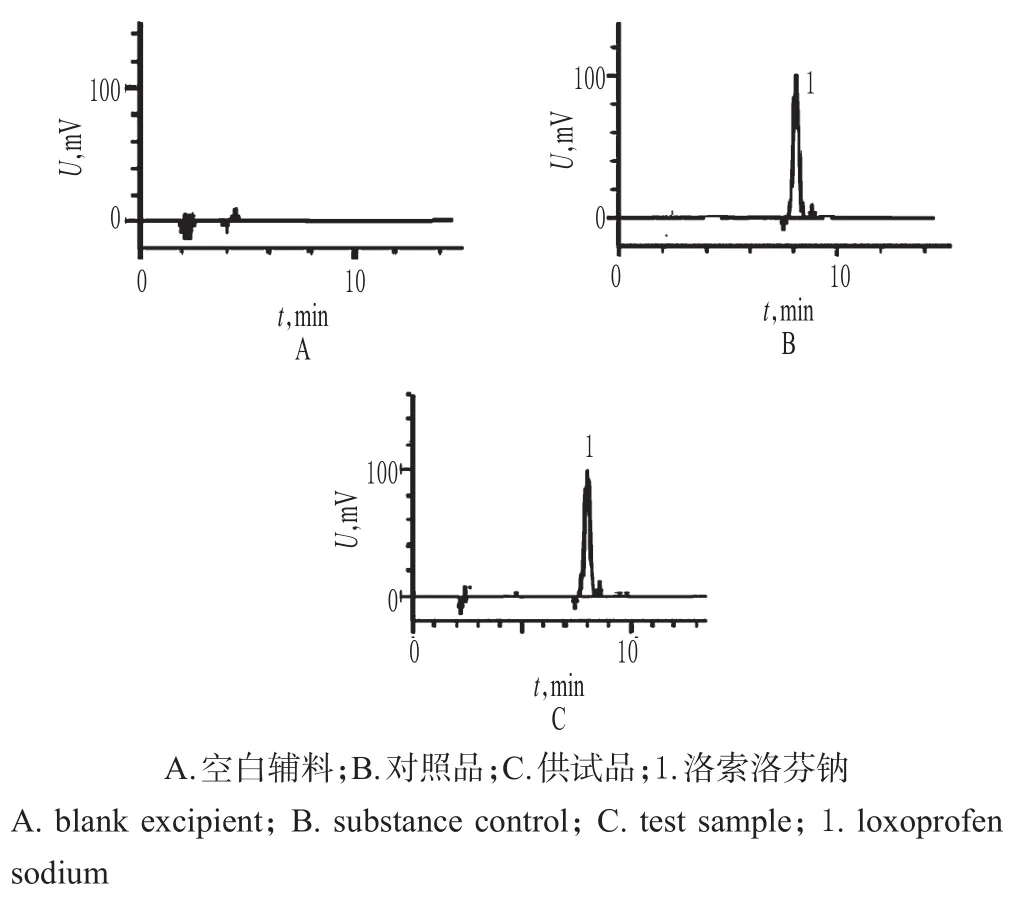
图1 系统适用性试验高效液相色谱图Fig 1 HPLC chromatogramsof system suitability tests
2.3.2 专属性试验 取样品10片(批号:110110),精密称定,研细,精密称取细粉适量(约相当于洛索洛芬钠30mg),加20m L的0.1mol/m L盐酸溶液,室温放置2 h后,用0.1mol/m L氢氧化钠溶液中和至中性,以流动相定容至50m L,作为酸破坏溶液;精密称取细粉适量(约相当于洛索洛芬钠30mg),加20m L的0.1mol/m L氢氧化钠溶液,室温放置2 h后,用0.1mol/m L盐酸溶液中和至中性,以流动相定容至50m L,作为碱破坏溶液;精密称取细粉适量(约相当于洛索洛芬钠30mg),加3%双氧水溶液10m L,室温放置4 h后,以流动相定容至50m L,作为氧化破坏溶液;精密称取细粉适量(约相当于洛索洛芬钠30mg),在60℃下放置5 d,以流动相定容至50 m L,作为热破坏溶液;精密称取细粉适量(约相当于洛索洛芬钠30mg),在(4 500±500)lx强光下照射5 d,以流动相定容至50m L,作为光破坏溶液。取上述各破坏试验溶液适量,分别按“2.1”项下色谱条件进样测定,记录色谱,详见图2。结果表明,虽经破坏后各杂质峰面积有一定增加,但洛索洛芬钠峰与相邻杂质峰之间、各杂质峰之间均能达到基线分离。
2.4 线性关系考察
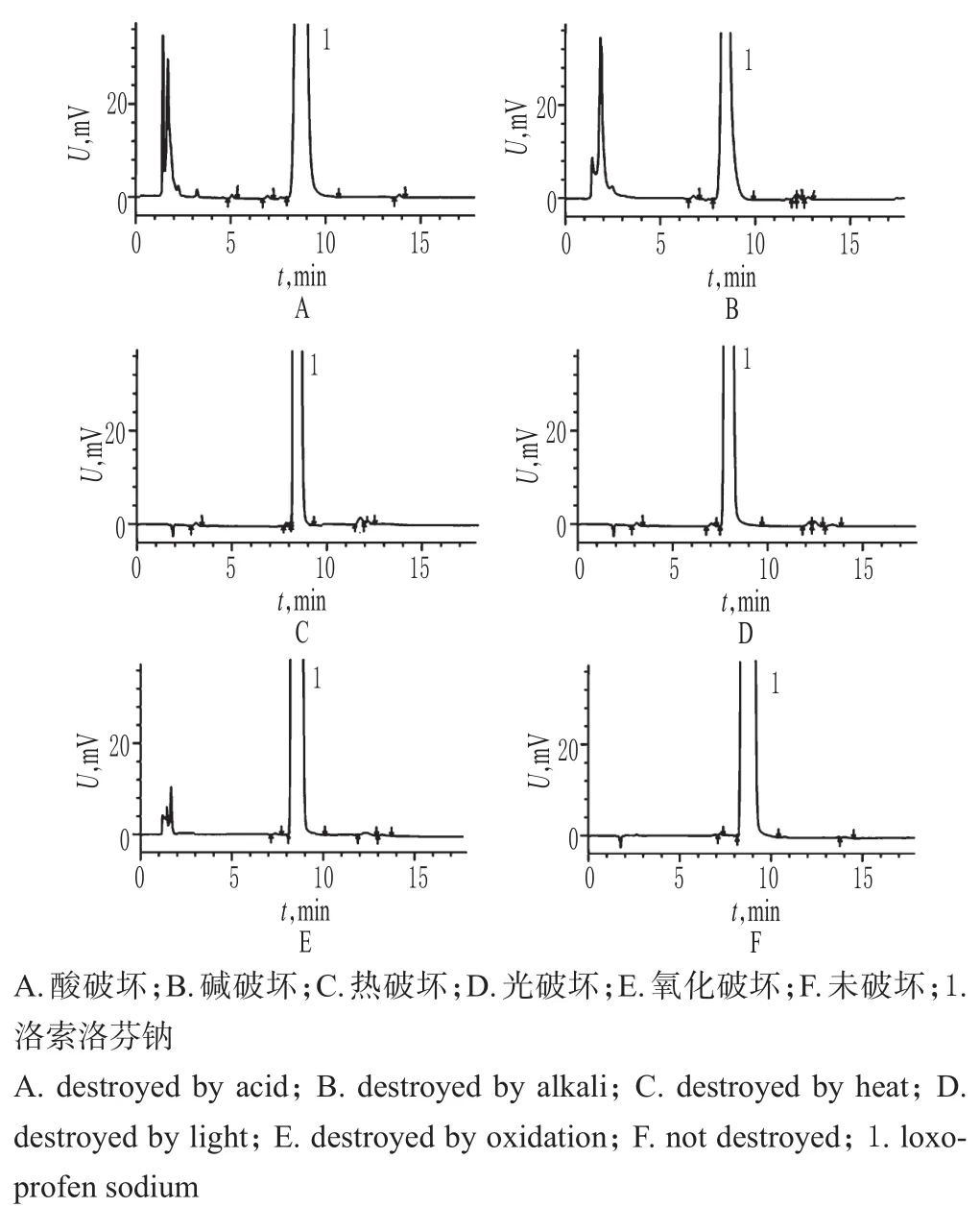
图2 专属性试验高效液相色谱图Fig 2 HPLC chromatogramsofspecificity tests
取洛索洛芬钠对照品30mg,精密称定,置于50m L量瓶中,加流动相溶解并稀释至刻度,摇匀,作为线性工作对照品贮备液。分别精密量取上述贮备液适量,用流动相稀释制成质量浓度为30.0、48.0、60.0、72.0、90.0μg/m L的系列线性工作对照品溶液,分别精密量取20μL,按“2.1”项下色谱条件进样测定,记录峰面积。以洛索洛芬钠峰面积(y)为纵坐标、质量浓度(x,μg/m L)为横坐标进行线性回归,得回归方程y=50 891x+3 043.5(r=0.999 8)。结果表明,洛索洛芬钠检测质量浓度线性范围为30.0~90.0μg/m L。
2.5 检测限
精密称取洛索洛芬钠对照品适量,用流动相制成每1m L中含0.3μg的溶液,精密量取20μL注入HPLC仪,按“2.1”项下色谱条件进样测定,记录峰面积。结果,此时信噪比约为3∶1,计算得到洛索洛芬钠的检测限为0.3μg/m L。
2.6 精密度试验
精密量取“2.2.2”项下对照品溶液适量,按“2.1”项下色谱条件连续进样测定6次,记录峰面积。结果,洛索洛芬钠峰面积的RSD=0.74%(n=6),表明仪器精密度良好。
2.7 稳定性试验
取“2.2.1”项下供试品溶液(批号:110110)适量,分别于室温下放置0、2、4、6、8、10 h时按“2.1”项下色谱条件进样测定,记录峰面积。结果,洛索洛芬钠峰面积的RSD=0.62%(n=6),表明供试品溶液室温下放置10 h内稳定性良好。
2.8 重复性试验
取同一批样品(批号:110110)适量,精密称定,分别按“2.2.1”项下方法平行制备6份供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算洛索洛芬钠含量。结果,洛索洛芬钠的平均标示百分含量为99.48%,RSD=0.57%(n=6),表明本方法重复性良好。
2.9 回收率试验
按样品处方精密称取空白辅料适量,共9份,每3份一组,分别精密加入低、中、高不同质量的洛索洛芬钠对照品,分别按“2.2.1”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,并计算洛索洛芬钠的回收率,结果见表1。

表1 回收率试验结果(n=9)Tab 1 Resultsof recovery tests(n=9)
2.10 样品含量及有关物质测定
2.1 0.1 样品含量测定 取3批样品各适量,分别按“2.2”项下方法制备对照品溶液和供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积,采用外标法以峰面积计算洛索洛芬钠含量,结果见表2。

表2 样品含量及有关物质测定结果(n=3)Tab 2 Resultsof contentsand related substancesdeterm ination of samples(n=3)
2.1 0.2 样品有关物质测定 每批样品取10片,精密称定,研细,精密称取细粉适量(约相当于洛索洛芬钠30 mg),置于100m L量瓶中,加流动相溶解并稀释至刻度,摇匀,滤过,取续滤液作为有关物质测定用供试品溶液;精密量取上述有关物质测定用供试品溶液适量,加流动相稀释制成每1m L中含洛索洛芬钠3μg的溶液,作为有关物质测定用对照溶液。按“2.1”项下色谱条件,取有关物质测定用对照溶液20μL注入HPLC仪,调节检测灵敏度,使主成分峰高约为满量程的25%;再取有关物质测定用供试品溶液20μL注入HPLC仪,记录色谱至主成分峰保留时间的2倍。供试品溶液色谱图中如显现除空白辅料峰外的杂质峰,则各杂质峰面积的总和不得大于对照溶液主峰面积的0.5倍(0.5%)。3批样品有关物质测定结果见表2。
3 讨论
洛索洛芬钠是第一个丙酸类前体型非甾体抗炎药,由日本三共株式会社首研,是目前日本销量第一的非甾体抗炎药。其经消化道吸收后迅速转化为trans-OH活性代谢物,通过抑制PG的生物合成而发挥作用,可明显减少胃肠道不良反应,且其镇痛效果比酮洛芬、吲哚美辛、萘普生强10~20倍。因此,该药具有较大的市场潜力。除了片剂,国内还曾见该药其他剂型如凝胶剂、贴剂等的研究。当前已有关于该药含量及有关物质测定、溶出度测定、生物利用度研究等方面的文献报道。《日本药局方》(第15版)[2-3]收录了洛索洛芬钠原料药和洛索洛芬钠片,采用液相色谱法(LC)对洛索洛芬钠的含量进行测定,采用薄层色谱法(TLC)对洛索洛芬钠的有关物质进行测定。此次笔者对自制的洛索洛芬钠片的含量及有关物质的测定进行了深入研究和改进,意在更全面地控制该制剂的质量。
以往文献报道的测定洛索洛芬钠含量的方法主要有紫外分光光度法[4]、HPLC法[5-6]和LC-质谱联用法(LCMS)[7],测定洛索洛芬钠有关物质的方法主要有RPHPLC法[8]。笔者在参考《日本药局方》(第15版)[主要色谱条件:流动相为甲醇-水-冰醋酸-三乙胺(60∶40∶0.1∶0.1,V/V/V/V),波长222 nm,柱温40℃]和原国家食品药品监督管理局国家药品标准[9]的基础上,改进了测定洛索洛芬钠含量及有关物质的RP-HPLC法。其中,经200~400 nm全波长范围内的紫外扫描考察,将测定波长调整为221 nm;同时,考虑到洛索洛芬钠呈弱碱性,流动相pH值和三乙胺浓度对其峰形和保留时间影响较大,通过对流动相不同组成、不同pH值、不同三乙胺浓度进行筛查,发现采用乙腈-0.01mol/L磷酸二氢钾溶液(含0.2%三乙胺,磷酸调节pH至3.0±0.1)(62∶38,V/V)作为流动相时,洛索洛芬钠的保留时间合适,且与其他相邻杂质峰的分离度符合要求,故确定将该流动相用于其含量及有关物质测定。
综上所述,本方法准确、简便、快速,适用于洛索洛芬钠片的质量控制。
[1] 周淑琴.新型非甾体抗炎药洛索洛芬钠的研究进展[J].上海医药,2008,29(10):468-469.
[2] The Society of Japanese Pharmacopoeia.The Japanese Pharmacopoeia[S].The fifteenth edition.Tokyo:Yakujinippo,Ltd.,2006:828.
[3] The Society of Japanese Pharmacopoeia.The Japanese Pharmacopoeia:SupplementⅡ[S].The fifteenth edition.Tokyo:Yakujinippo,Ltd.,2006:2713.
[4] 唐黎明,陈桂良.洛索洛芬钠片溶出度测定及体内外相关性评价[J].中国医药工业杂志,2006,37(8):550-552.
[5] 冀满丰.HPLC法测定洛索洛芬钠缓释片中主药的含量[J].中国现代药物应用,2008,2(12):8-9.
[6] 罗静,张涛,黄华,等.洛索洛芬钠巴布剂的制备及质量评价[J].中国医药工业杂志,2012,43(2):107-111.
[7] 张蓓蓓,张尊建,田媛,等.洛索洛芬钠缓释片在比格犬体内的药代动力学[J].中国药科大学学报,2006,37(4):333-336.
[8] 陆步实,张根元,杨大龙,等.反相高效液相色谱法测定洛索洛芬钠的含量及其有关物质[J].中国新药杂志,2004,13(12):1137-1139.
[9] 国家食品药品监督管理局.洛索洛芬钠国家药品标准:WS1-(X-137)-2005Z[S].2005.
(编辑:周 箐)
Determination of Contentsand Related Substances in Loxoprofen Sodium Tabletsby RP-HPLC
YUAN Haoyu,WANG Peng,YIHong,YU Yu(No.416 Hospital of Nuclear Industry,Chengdu 610051)
OBJECTIVE:To improve the determ ination method for the contents of main components and related substances in Loxoprofen sodium tablets.METHODS:RP-HPLC method was adopted.The determ ination was performed on Inspire C18column w ith mobile phase consisted of acetonitile-0.01 mol/L potassium dihydrogen phosphate(containing 0.2%triethylamine,phosphoric acid adjusted to 3.0±0.1,62∶38,V/V)at the flow rate of 1.0 m L/m in.The column temperature was 40℃,and the detection wavelength was set at 221 nm.The sample size was 20μL.RESULTS:The peak of loxoprofen sodium was well separated w ith the peak of its related substances(R>1.5).The linear range of loxoprofen sodium ranged 30.0-90.0μg/m L(r=0.999 8).The detection lim it of loxoprofen was 0.3μg/m L.RSDs of precision,stability and repeatability tests were<1.0%.The average recovery rates ranged 99.00%-99.87%(RSD=0.33%,n=9).CONCLUSIONS:Thismethod is accurate,simple,rapid and suitable for the quality control of Loxoprofen sodium tablets.
RP-HPLC;Loxoprofen sodium tablets;Content;Related substances
R927
A
1001-0408(2017)15-2127-04
2016-05-31
2017-04-06)
*副主任药师,硕士。研究方向:医院药学。E-mail:yhy416@ 126.com
#通信作者:副主任药师。研究方向:医院药学。E-mail:yihon g0517@163.com
DOI 10.6039/j.issn.1001-0408.2017.15.31
