HPLC法测定注射用泮托拉唑钠中的有关物质
2017-07-07翟丽杰高立娜吉林大学第二医院药学部长春130041
张 静,翟丽杰,高立娜(吉林大学第二医院药学部,长春 130041)
HPLC法测定注射用泮托拉唑钠中的有关物质
张 静*,翟丽杰#,高立娜(吉林大学第二医院药学部,长春 130041)
目的:建立测定注射用泮托拉唑钠中有关物质的方法。方法:采用高效液相色谱法。色谱柱为KromasilHypersilODS,流动相为0.01mol/L磷酸氢二钾溶液(调节pH为7.0)-乙腈(梯度洗脱),流速为1.0m L/m in,检测波长为290 nm,柱温为40℃,进样量为20μL。结果:杂质A、B、C+E、D检测质量浓度线性范围分别为0.416 8~1.042 0μg/m L(r=0.999 8)、0.195 0~0.487 5μg/m L(r=0.999 9)、0.389 0~0.972 5μg/m L(r=0.999 8)、0.198 6~0.496 5μg/m L(r=0.999 8);定量限分别为0.834、0.780、1.556、0.794 ng/m L,检测限分别为0.417、0.390、0.778、0.397 ng/m L;精密度试验的RSD<1.0%,重复性试验中总杂质峰面积的RSD<1.0%;回收率分别为98.81%~102.49%(RSD=1.18%,n=9)、95.31%~98.44%(RSD=0.91%,n=9)、96.88%~98.44%(RSD=0.52%,n=9)、97.87%~101.28%(RSD=1.05%,n=9)。结论:该方法简便、准确,可用于注射用泮托拉唑钠中有关物质的测定。
高效液相色谱法;注射用泮托拉唑钠;有关物质;测定
泮托拉唑钠由德国百克顿(Gulden)公司开发上市,是继奥美拉唑、兰索拉唑之后的新一代质子泵抑制剂(PPI),为消化性溃疡的一线治疗药[1-6]。在全球上市后,因其抑酸作用强而持久、溃疡愈合率高、不良反应少和药物相互作用少的显著优势,迅速成为治疗与胃酸有关的消化系统功能紊乱性疾病的特效药物。其与奥美拉唑和兰索拉唑相比,对质子泵具有更高的选择性,抑制胃酸的分泌达到一个新水平,疗效明显占优。目前,该制剂已被新版《中国药典》[7]、《英国药典》[8]、《美国药典》[9]和《欧洲药典》[10]收载。而《英国药典》《美国药典》《欧洲药典》收载的仅有泮托拉唑钠原料药标准,无注射用无菌粉末标准;而《中国药典》中虽收载了泮托拉唑钠原料药和注射用无菌粉末标准,但是并没有对各杂质作相应的限度要求。因此,本研究参考相关文献[7],采用高效液相色谱法(HPLC)建立了测定注射用泮托拉唑钠中有关物质的方法,以期为完善该制剂的质量标准提供参考。
1 材料
1.1 仪器
LC-20A型HPLC仪,包括LC-20AD XR二元梯度泵、SIL 20A HT自动进样器、CTO-20A柱温箱、SPDM 20A二极管阵列检测器(日本Shimadzu公司);FE20K型酸度计(瑞士Mettler-Toledo公司);ZH-2型旋涡混合器(天津药典标准仪器厂);M illi-Q Advantage A10型超纯水仪(美国M illipore公司)。
1.2 药品与试剂
注射用泮托拉唑钠[石药集团中诺药业(石家庄)有限公司,批号:C058131101、C058131102、C058131103,规格:60mg/支];注射用泮托拉唑钠(原研药,德国奈科明制药有限公司,批号:203869,规格:40mg/支);泮托拉唑钠对照品(批号:20140511,纯度:99%)、杂质A对照品(批号:20140511,纯度:99%)、杂质B对照品(批号:20140511,纯度:99%)、杂质C对照品(批号:20140511,纯度:99%)、杂质E对照品(批号:20140511,纯度:99%)均购自深圳达尔森科技有限公司;杂质D对照品(美国Waters公司,批号:20140511,纯度:99%);乙腈为色谱纯,其余试剂均为分析纯,水为超纯水。
2 方法与结果
2.1 色谱条件
色谱柱:Kromasil Hypersil ODS(125mm×4.0mm,5µm);流动相:0.01mol/L磷酸氢二钾溶液(调节pH为7.0)(A)-乙腈(B),梯度洗脱(0~40min,80%→20%A;40~45min,20%→80%A);流速:1.0m L/m in;检测波长:290 nm;柱温:40℃;进样量为20µL。
2.2 溶液的制备
2.2.1 混合对照品溶液 精密称取泮托拉唑钠对照品和杂质A、B、C、D、E对照品各约1mg,分别置于10m L量瓶中,加乙腈溶解并定容,摇匀,即得泮托拉唑钠和杂质A、B、C、D、E的单一对照品贮备液。分别量取上述泮托拉唑钠单一对照品贮备液1m L,杂质A单一对照品贮备液80μL,杂质B、C、D、E单一对照品贮备液各40μL,置于同一10m L量瓶中,加乙腈溶解并定容,摇匀,即得混合对照品溶液。
2.2.2 供试品溶液 取样品适量,置于25m L量瓶中,加流动相溶解并定容,摇匀,经0.45μm微孔滤膜滤过,取续滤液,即得。
2.2.3 空白对照溶液 取处方比例辅料适量(约相当于泮托拉唑钠4mg处方所需辅料量),置于100m L量瓶中,加流动相溶解并定容,摇匀,经0.45μm微孔滤膜滤过,取续滤液,即得。
2.2.4 系统适用性溶液 取杂质A、杂质B、杂质C、杂质D、杂质E、泮托拉唑钠对照品各适量,置于5m L量瓶中,加流动相溶解并定容,摇匀,即得。
2.3 系统适用性和专属性试验
取“2.2”项下混合对照品溶液、供试品溶液、空白对照溶液、系统适用性溶液各适量,按“2.1”项下色谱条件进样测定,记录色谱,详见图1。由图1可知,在该色谱条件下,各待测有关物质均能达到基线分离,分离度>1.5;理论板数以泮托拉唑钠峰计为15 800,保留时间为11.025min。结果表明,其他成分对测定无干扰。
2.4 破坏性试验

图1 系统适用性和专属性试验高效液相色谱图Fig 1 HPLC chromatograms of system suitability and specificity test
按如下方法制备各破坏样品溶液——(1)酸破坏样品溶液:取样品适量(约相当于泮托拉唑钠40mg),置于100m L量瓶中,加0.01mol/L盐酸溶液12m L,于室温下静置10min,加适量0.01mol/L氢氧化钠溶液中和,再加0.001mol/L氢氧化钠溶液-乙腈(1∶1,V/V)溶解并定容,摇匀,经0.45μm微孔滤膜滤过,取续滤液,即得。(2)碱破坏样品溶液:取样品适量(约相当于泮托拉唑钠40mg),置于100m L量瓶中,加1mol/L氢氧化钠溶液12m L,振摇使其全部溶解,于室温下放置1 h,加适量1mol/L盐酸溶液中和,再加0.001mol/L氢氧化钠溶液-乙腈(1∶1,V/V)溶解并定容,摇匀,经0.45μm微孔滤膜滤过,取续滤液,即得。(3)高温破坏样品溶液:取样品适量(约相当于泮托拉唑钠40mg),置于圆皿内,于120℃加热40min,取出,冷却至室温,置于100m L量瓶中,加0.001mol/L氢氧化钠溶液-乙腈(1∶1,V/V)溶解并定容,摇匀,经0.45μm微孔滤膜滤过,取续滤液,即得。(4)高湿破坏样品溶液:取样品适量(约相当于泮托拉唑钠40mg),置于平皿内,开口置于盛有硝酸钾饱和溶液的密闭容器中(湿度为92.5%),于25℃条件下放置72 h,置于100m L量瓶中,加0.001mol/L氢氧化钠溶液-乙腈(1∶1,V/V)溶解并定容,摇匀,经0.45μm微孔滤膜滤过,取续滤液,即得。(5)光照破坏样品溶液:取样品适量(约相当于泮托拉唑钠100mg),置于平皿内,开口置于(7 500±500)lx强光下照射72 h,置于100m L量瓶中,加0.001mol/L氢氧化钠溶液-乙腈(1∶1,V/V)溶解并定容,摇匀,经0.45μm微孔滤膜滤过,取续滤液,即得。
取上述破坏样品溶液各适量,按“2.1”项下色谱条件进样测定,记录色谱,详见图2。由图2可知,样品在酸、碱、高温、高湿、光照条件下均有不同程度的降解,杂质峰有所增多,但各降解杂质峰与主成分峰之间以及各降解杂质峰之间分离良好(分离度均>1.5)。
2.5 线性关系考察
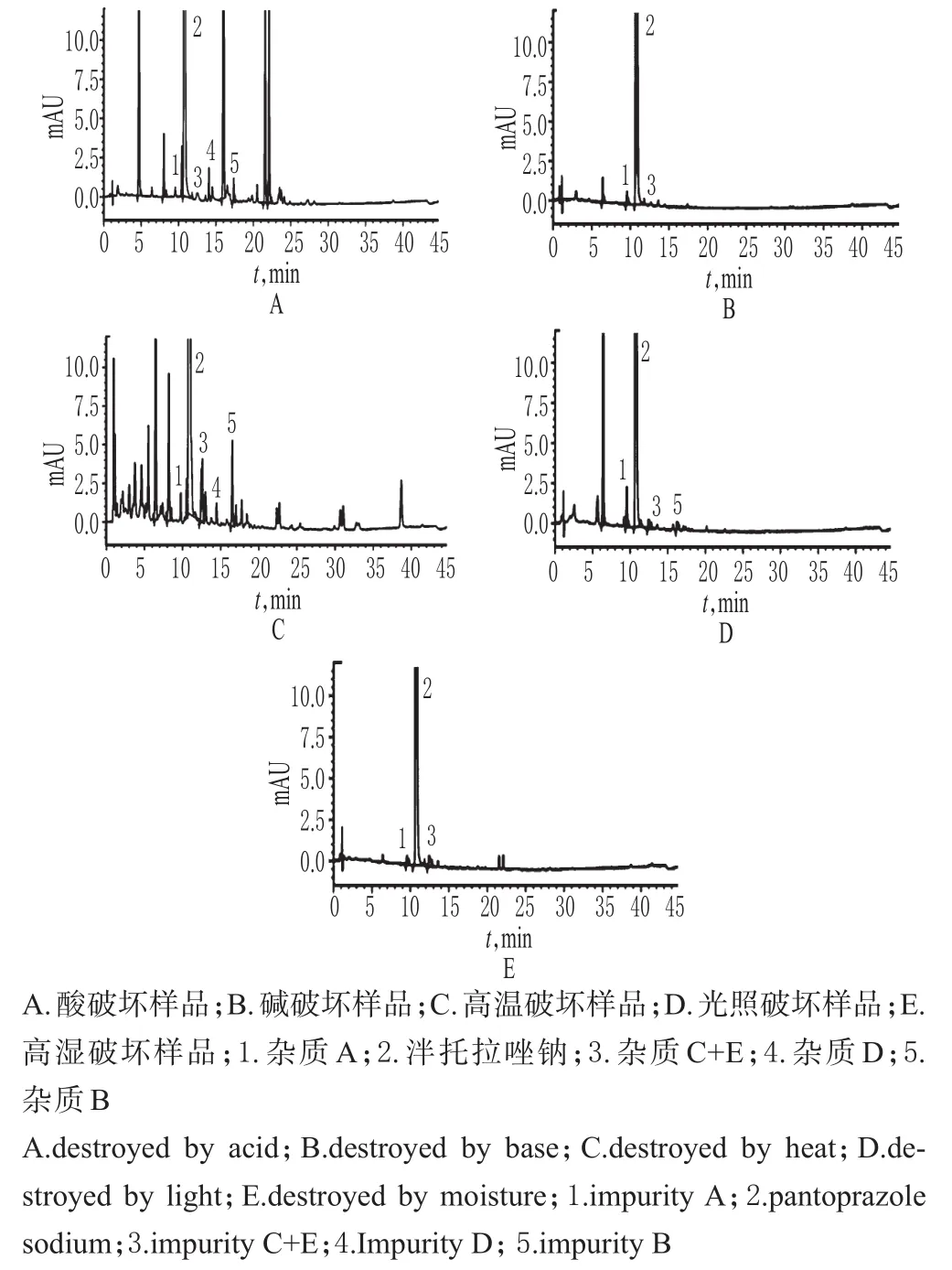
图2 破坏性试验高效液相色谱图Fig 2 HPLC chromatogramsofdestructive test
范围见表1。

表1 回归方程与线性范围Tab 1 Regression equationsand linear ranges
2.6 定量限与检测限考察
取“2.2.1”项下混合对照品溶液适量,倍比稀释,按“2.1”项下色谱条件连续进样测定6次,记录峰面积。当信噪比为10∶1时,得杂质A、B、C+E、D的定量限(LOQ)分别为0.834、0.780、1.556、0.794 ng/m L;当信噪比为3∶1时,得检测限(LOD)分别为0.417、0.390、0.778、0.397 ng/m L。
2.7 精密度试验
取“2.2.1”项下混合对照品溶液适量,按“2.1”项下色谱条件连续进样测定6次,记录峰面积。结果,杂质A、B、C+E、D峰面积的RSD分别为0.03%、0.03%、0.06%、0.05%(n=6),表明仪器精密度良好。
2.8 重复性试验
取样品(批号:C058131101)适量,共6份,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积。结果,供试品溶液中只检出杂质A和杂质C+E,杂质A峰面积的RSD=0.22%(n=6),杂质C+E峰面积的RSD=0.17%(n=6),总杂质峰面积的RSD=0.98%(n=6),表明本方法重复性良好。
2.9 回收率试验
从联盟初创期特征来看,各BIM联盟在分析内外部条件基础上,确定各自战略目标、寻找合作伙伴,签署联盟协议和纲领文件,设立专家库、成立专家委员会,制定发展规划、技术路线,开展标准研究制定、课题研究、人才培养、互动交流等工作。但对于联盟技术突破性创新、全产业链深度合作、利益分配共享、风险共担、资金投入、市场占领等内容并未充分涉及。
分别量取“2.2.1”项下杂质A、B、C、D、E的单一对照品贮备液各适量,置于同一10 m L量瓶中,加0.001 mol/L氢氧化钠溶液-乙腈(1∶1,V/V)溶解并定容,摇匀,即得。按上述方法制备低、中、高质量浓度的混合对照品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算回收率,结果见表2。
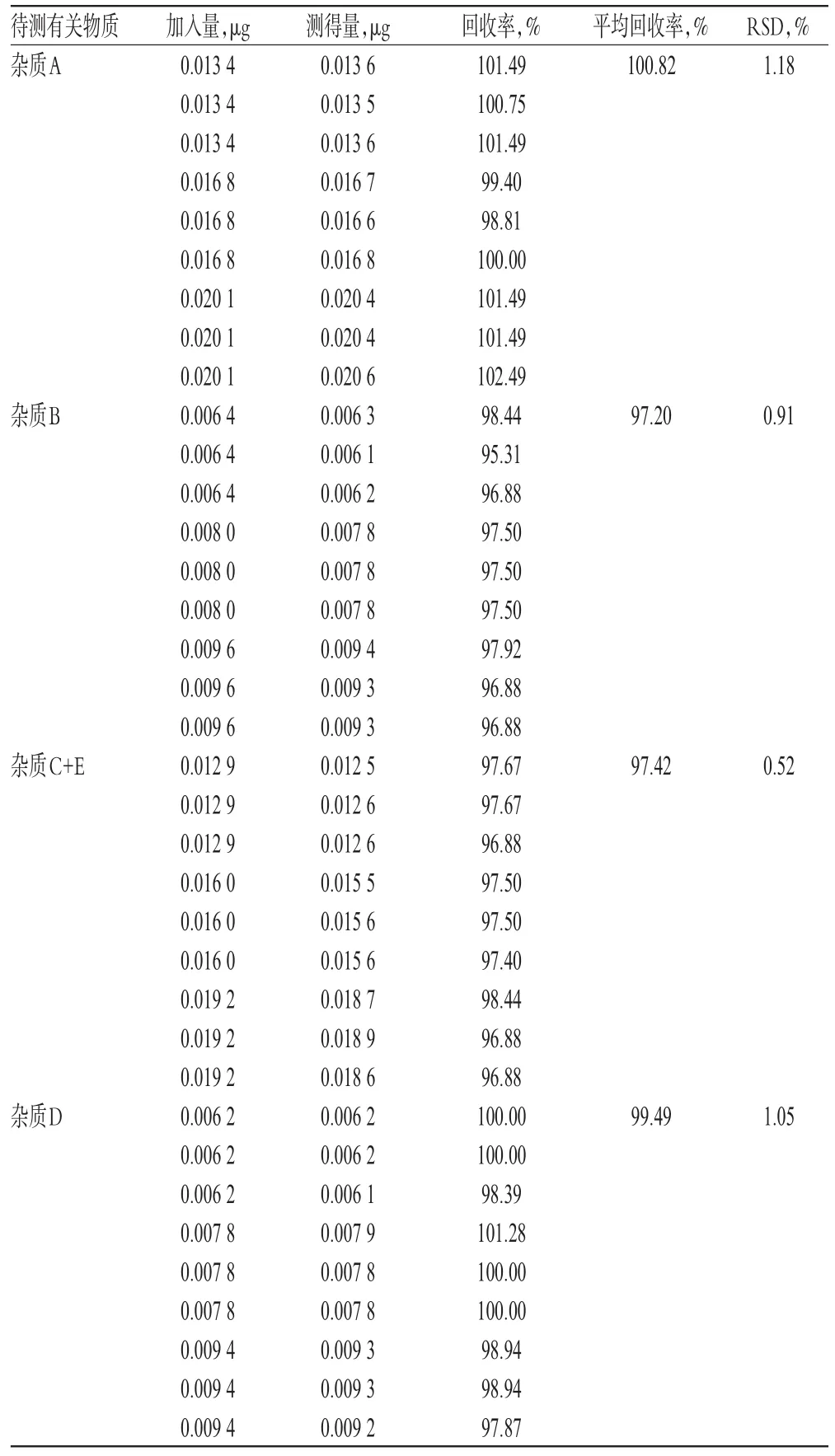
表2 回收率试验结果(n=9)Tab 2 Resultsof recovery test(n=9)
2.10 样品有关物质测定
取4批样品各适量,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算样品中含有关物质的量,结果见表3。由表3可知,3批国产样品均只检测出两个已知杂质,即杂质A以及杂质C+E,并均<0.2%,总杂质<1.0%;原研药样品中杂质C+E含量>0.2%,杂质A未达到检测限。
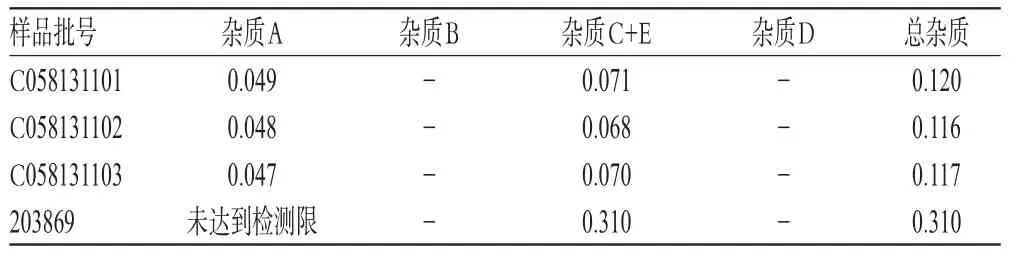
表3 样品有关物质测定结果(n=3,%)Tab 3 Resultsof related substancesdeterm ination of samples(n=3,%)
3 讨论
3.1 柱温的选择
笔者考查了不同柱温(30℃和40℃)对样品分离的影响。结果表明,不同柱温的测试结果均符合系统适用性要求,保留时间无明显差别;但柱温40℃条件下各峰对称性、分离度及柱效更好,更有利于样品的分离。因此,本试验的柱温选择为40℃。
3.2 流动相A的pH考察
笔者考查了流动相A的不同pH(6.5、7.0和7.5)对样品中已知杂质分离的影响。结果表明,流动相A的pH为6.5时,杂质A与主峰无法分离;流动相A的pH为7.5时,杂质C+E与杂质D无法完全分离;而流动相A的pH为7.0时,各杂质与主峰以及各杂质间的分离均较好。故本试验选择流动相A的pH为7.0。
3.3 稳定性试验
本试验曾进行稳定性试验,考察了供试品溶液(批号:C058131101)在室温下放置0、1、2、3、4 h时的总杂质峰面积。结果,总杂质峰面积在4 h内明显增加,表明进行有关物质测定时,供试品溶液应在制备后立即测定,以免对测定结果产生不必要的干扰。
3.4 杂质C+E的考察
注射用泮托拉唑钠中的杂质C为5-(二氟甲氧基)-2-{(RS)-[(3,4-二甲氧基-2-吡啶基)甲基]亚磺酰基}-1甲基-1H-苯骈咪唑,杂质E为6-(二氟甲氧基)-2-{(RS)-[(3,4-二甲氧基-2-吡啶基)甲基]亚磺酰基}-1甲基-1H-苯骈咪唑,杂质C是以杂质B(硫醚)为起始原料经过氧化物氧化得到的,其与杂质E互为异构体,无法完全分离[9-10]。本文参考《欧洲药典》和《美国药典》系统适用性标准[9-10],测定杂质C和E时,以其总量C+E表示,作为同一杂质考虑。
综上所述,本方法简便、准确,可用于注射用泮托拉唑钠中有关物质的测定。
[1] 李军,部敬顺,张鉴.注射用泮托拉唑钠的稳定性考察[J].中国药房,2005,16(21):1655-1657.
[2] 孙丹,朱娴.注射用泮托拉唑钠在各注射液中配伍的稳定性考察[J].化工中间体,2015(10):114-115.
[3] 王丽云,吕旭幸.注射用泮托拉唑钠的杂质含量及其杂质谱的比较[J].抗感染药学,2015,12(4):499-504.
[4] 蒋鹏,钱新毅.质子泵抑制药:泮托拉唑[J].医药导报,2003,22(3):184-186.
[5] 王婧斯,李桂龙,王成港,等.泮托拉唑钠有关物质分析方法的比较[J].药物评价研究,2012,35(2):109-112.
[6] 王荔,毕煌垒.泮托拉唑不良反应分析[J].中国药房,2010,21(28):2582-2683.
[7] 国家药典委员会.中华人民共和国药典:二部[S].2015年版.北京:中国医药科技出版社,2015:704-706.
[8] British Pharmacipoeia Comm ission Office.British Pharmacopoeia 2015:VolumeⅡ[S].2015:494.
[9] The United Stated Pharmacipoeia Commission Office.The United Stated Pharmacopoeia:37[S].2015:3264.
[10] European Pharmacipoeia Comm ission Office.European Pharmacopoeia:8.0[S].2015:4680.
(编辑:刘 柳)
Determ ination of Related Substances in Pantoprazole Sodium for Injection by HPLC
ZHANG Jing,ZHAILijie,GAO Lina(Dept.of Pharmacy,the Second Hospital of Jinlin University,Changchun 130041,China)
OBJECTIVE:To establish a method for the determination of related substances in Pantoprazole sodium for injections.METHODS:HPLC method was adopted.The determination was performed on Kromasil Hypersil ODS column w ith mobile phases consisting of 0.01 mol/L potassium dihydrogen phosphate buffer solution(pH adjusted to 7.0)-acetonitrile(gradient elution)at a flow rate of 1.0 m L/m in.The detection wavelength was set at 290 nm,and the column temperature was 40℃,and injection volume was 20μL.RESULTS:The linear ranges of impurity A,impurity B,impurity C+E,and impurity D were 0.416 8-1.042 0 μg/m L(r=0.999 8),0.195 0-0.487 5μg/m L(r=0.999 9),0.389 0-0.972 5μg/m L(r=0.999 8),0.198 6-0.496 5μg/m L(r=0.999 8),respectively.The limits of quantitation were 0.834,0.780,1.556,0.794 ng/m L;the limits of detection were 0.417,0.390,0.778,0.397 ng/m L,respectively.RSD of precision testwas lower than 1.0%;in repetitive test,RSD for total peak area of impurity was lower than 1.0%;the recoveries were 98.81%-102.49%(RSD=1.18%,n=9),95.31%-98.44%(RSD=0.91%,n=9),96.88%-98.44%(RSD=0.52%,n=9)and 97.87%-101.28%(RSD=1.05%,n=9).CONCLUSIONS:Themethod is convenient,accurate and suitable for the determ ination of related substance in Pantoprazole sodium for injection.
HPLC;Pantoprazole sodium for injection;Related substances;Determ ination
R927
A
1001-0408(2017)15-2142-04
2016-08-18
2016-12-19)
*主管药师,硕士。研究方向:临床药学。电话:0431-88796629。E-mail:jingwei_zhang@sina.com
#通信作者:副主任药师,硕士。研究方向:临床药学。电话:0431-88796413。E-mail:109788809@qq.com
DOI 10.6039/j.issn.1001-0408.2017.15.35
