过渡元素掺杂Fe3O4(001)表面磁电性能的理论研究
2017-07-05赵中霞任韧任奕璟周志立
赵中霞 任韧 任奕璟 周志立
(西安交通大学理学院,西安710049)
过渡元素掺杂Fe3O4(001)表面磁电性能的理论研究
赵中霞 任韧*任奕璟 周志立
(西安交通大学理学院,西安710049)
采用基于密度泛函理论的第一性原理方法,计算Fe3O4,Fe3O4(001)表面以及过渡元素掺杂表面的电子结构和磁性。结果表明Fe3O4的半金属性主要来源于B位Fe离子,并且Fe的3d轨道发生强烈自旋极化;比较(001)表面不同终端A和B终端的表面能和电子结构,得出两种终端稳定性存在差异且A终端较稳定同时表现半金属性;由过渡元素V、Cr、Mn、Co、Cu和Zn取代Fe3O4(001)表面A终端A位Fe进行掺杂,形成的6种新表面结构都保持了半金属性。对比它们的表面能和磁矩,Mn掺杂的表面结构最稳定并且磁矩明显增大。
Fe3O4;密度泛函理论;电子结构;磁性能;表面能
0 引言
早在1983年,De Groot等[1]对half-Heusler合金NiMnSb和PtMnSb等材料的电子结构进行研究时,发现一种新型的特殊能带结构。这种能带结构同时具有金属和绝缘体的能带特性,即一种自旋方向的电子态密度越过费米能级,此方向的电子导电,表现金属性;而另一种自旋方向的电子,费米能级处于能隙内,此方向的电子不导电,具有这种能带结构的物质称为半金属。半金属的特殊能带结构,使其具备了特殊的性质:它在费米能级的电子自旋极化率理论上高达100%[2-4];它的磁矩是玻尔磁矩的整数倍[3,5];某些半金属具有很高的居里温度[2,6]。半金属的这些特殊性质,使其在核自旋压力显微成像和自旋电子器件中具有非常广泛的应用前景。此后,在对半金属的系统研究中,人们已经发现了数百种半金属材料。其中,亚铁磁性氧化物Fe3O4表现出优异的半金属性[7],电子自旋极化率大。对亚铁磁性半金属Fe3O4以及Fe3O4表面的理论和实验研究引起学者关注[8-13]。Szotek等[10]基于第一性原理对Fe3O4的磁电性能包括电子结构、电荷有序性等进行了理论研究;随后,为了改进Fe3O4的磁电性能,并探索更多性能优异的新型半金属材料,Szotek等[11]采用过渡元素Fe、Co和Ni对Fe3O4进行掺杂得到XFe2O4(X=Fe,Co,Ni),并计算分析了它们的能带结构,电子分布以及磁矩等磁电性能;Spiridis等[12-13]分别通过扫描隧道显微镜和密度泛函理论研究,发现了Fe3O4的(001)表面模型的A层终端;Mariotto等[14]提出了Fe3O4的(001)表面模型的B层终端。Yu等[15]基于密度泛函理论详细计算分析了Fe3O4(111),(110)以及(001)表面的电子结构,稳定性以及磁性能。
但是,尚没有对Fe3O4和Fe3O4表面的磁电性能以及对表面掺杂后得到的新表面结构的磁电性能进行系统研究。本文运用第一性原理计算方法,通过密度泛函理论[16],对Fe3O4的电子结构,分子磁矩等性能进行详细计算和分析。并计算和比较Fe3O4(001)表面的两个不同终端模型的表面结构,表面能,电子结构和总磁矩,得到最稳定的表面模型。采用过渡元素V,Cr,Mn,Co,Cu和Zn对Fe3O4(001)表面A层终端进行掺杂得到新的表面结构,并分析它们的表面能,电子结构和分子磁矩等半金属性能,为获得更多具有优异性能的新型半金属材料提供了理论依据。
1 模型和计算方法
1.1 Fe3O4结构模型和计算方法
反尖晶石结构Fe3O4的晶体结构[4]为A位Fe离子占据O形成的四面体间隙,B位Fe离子占据O形成的八面体间隙,FeA-O键长是0.188 6 nm,FeB-O键长是0.205 7 nm;图1(a)和(b)是反尖晶石结构Fe3O4的晶胞和原胞,(c)和(d)分别是A位和B位Fe离子。在这里,以Fe3O4的原胞为计算对象,通过基于密度泛函理论[17-18]模块进行计算。结构优化时,精度收敛采用超精细,即能量变化不超过5.0×10-6eV·atom-1,原子间相互作用力不超过0.10 eV·nm-1,最大位移不超过5.0×10-5nm。自洽迭代k点设置,采用MonkhorstPack[19]取样,使用444个网格。使用赝势平面波展开波函数,原子核与价电子之间的相互作用采用超软赝势[20-21],截断能设470 eV。由于Fe3O4包含d电子,存在电子间强关联效应,在采用广义梯度近似(GGA)[22]处理交换关联能时,需要加入Hubbard参数[23],这里Hubbard U值分别取U=0~7 eV进行计算,分析计算结果,从中选出最合理的U值,从而进一步对Fe3O4的电子结构和磁性进行应用分析[33-35]。
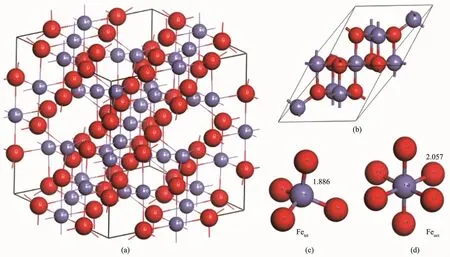
图1 (a)Fe3O4晶胞;(b)Fe3O4的原胞;(c)A位Fe离子;(d)B位Fe离子(红色是O,蓝色是Fe)Fig.1(a)Conventional cell of Fe3O4;(b)Primitive cell of Fe3O4;(c)Fe-ions on A-sites; (d)Fe-ions on B-sites(Red for O,blue for Fe)
1.2 Fe3O4(001)表面模型和计算方法
Fe3O4(001)表面是由A层和B层交替重叠堆积而成,其中A层终端包含四面体间隙Fe,B层终端包含八面体间隙Fe和O,如图2。在这里设置9层原子面进行模拟,并且固定中间3层原子面,真空层厚度设为1.5 nm。仍然采用上述计算方法优化并计算Fe3O4(001)表面2个不同终端的表面结构,表面能,电子结构和磁性。表面能的计算公式为[24]:
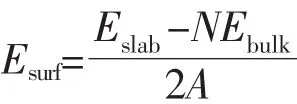
其中,Eslab是表面模型的能量,N是slab中的单胞数,Ebulk是一个单胞的能量,A是表面积。通过分别计算2个终端的表面能,来比较它们的稳定性。
1.3 Fe3O4(001)表面掺杂模型和计算方法
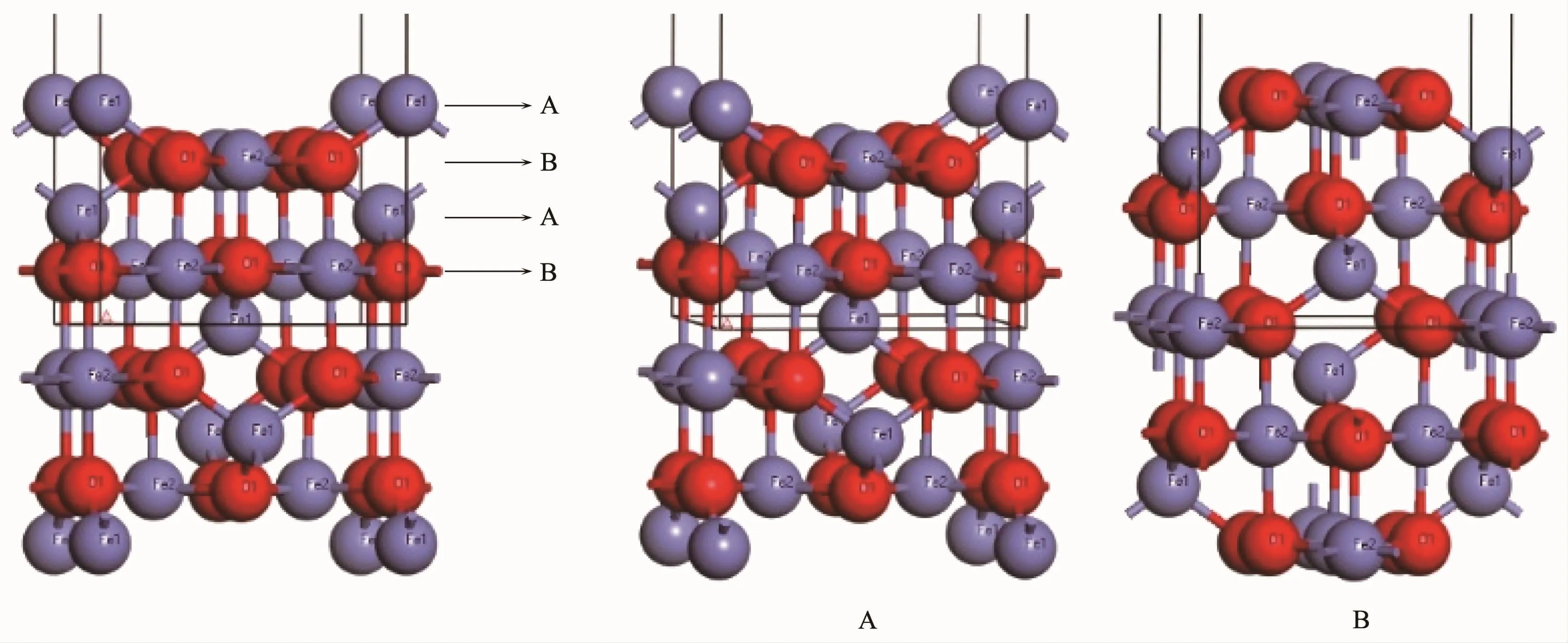
图2 Fe3O4(001)表面及2个终端Fig.2Fe3O4(001)plane and its two terminations
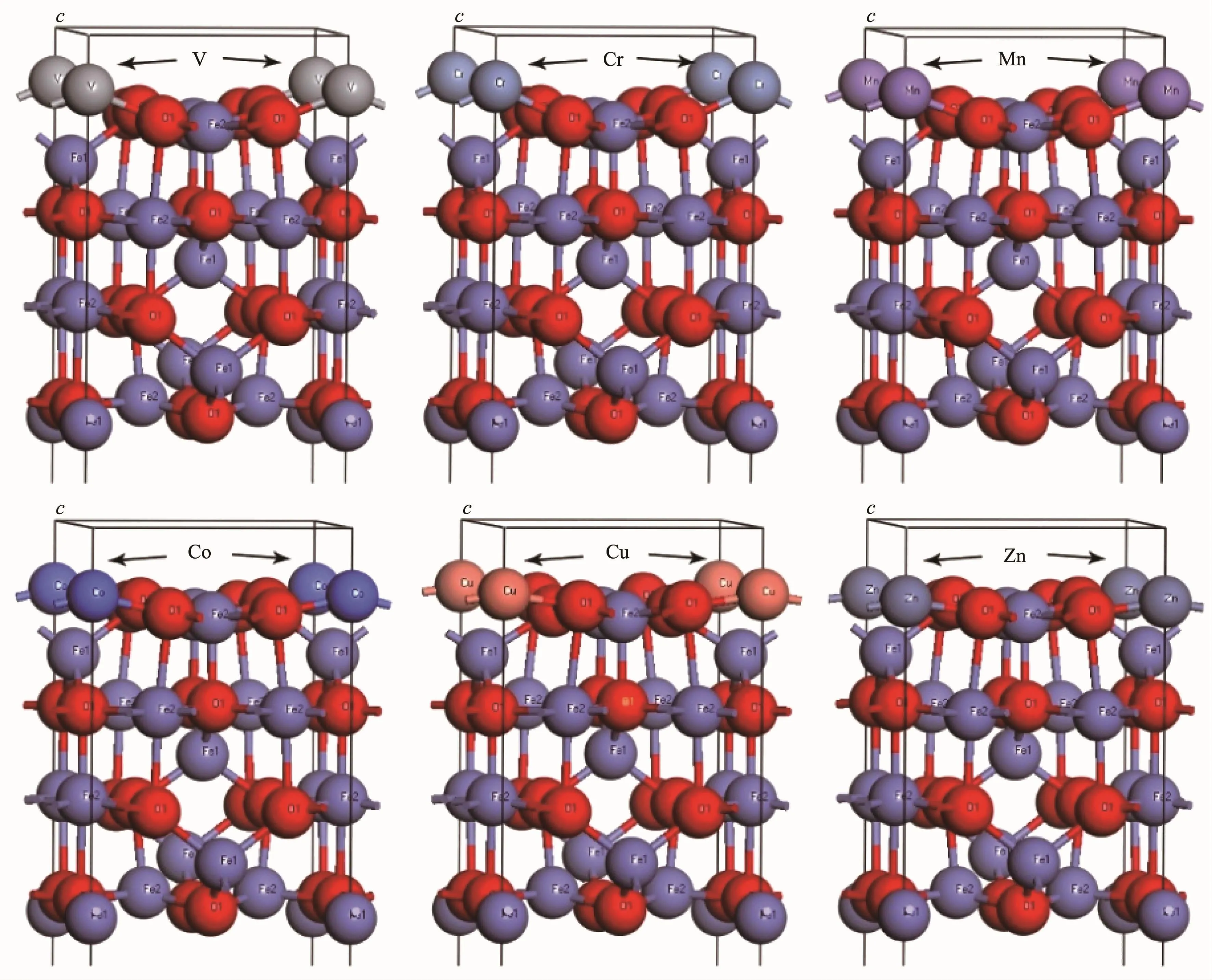
图3 Fe3O4(001)表面A终端第一层FeA被V、Cr、Mn、Co、Cu和Zn取代Fig.3FeAof Fe3O4(001)plane were substituted by V,Cr,Mn,Co,Cu and Zn
选取Fe3O4(001)表面更稳定的A层终端进行表面掺杂,如图3所示分别用过渡元素V、Cr、Mn、Co、Cu和Zn取代Fe3O4(001)表面A层终端第一层的A位Fe形成新表面材料,在这里设置9层原子面进行模拟,真空层厚度设为1.5 nm,并且固定中间3层原子面。同样采用上述方法,分别详细计算和分析各个新表面的表面能,态密度,电荷密度和磁矩等磁电性能,并进行比较。
2 结果与讨论
2.1 Fe3O4的电子结构和磁性
采用GGA+U的方法对强关联体系Fe3O4进行计算,图4中黑线(实线)代表Hubbard U值分别取0,1,2,3,3.5,4,5,6和7 eV时计算得到的Fe3O4分子磁矩,红线(虚线)表示Fe3O4分子磁矩的实验值4.1μB[25],可以看出,当U取3.5 eV时,得到的分子磁矩4.02μB与实验值4.1μB最为接近,因此,得到计算过程中的Hubbard U值取3.5 eV。当U=3.5 eV时计算得到的带隙是0.35 eV,与Yu等[15]计算得到的带隙值接近,进一步验证了U值的合理性。同时,在此U值下,优化后得到的晶格常数是0.838 6 nm,与实验值0.839 nm以及理论计算值0.841 nm接近[26-27]。
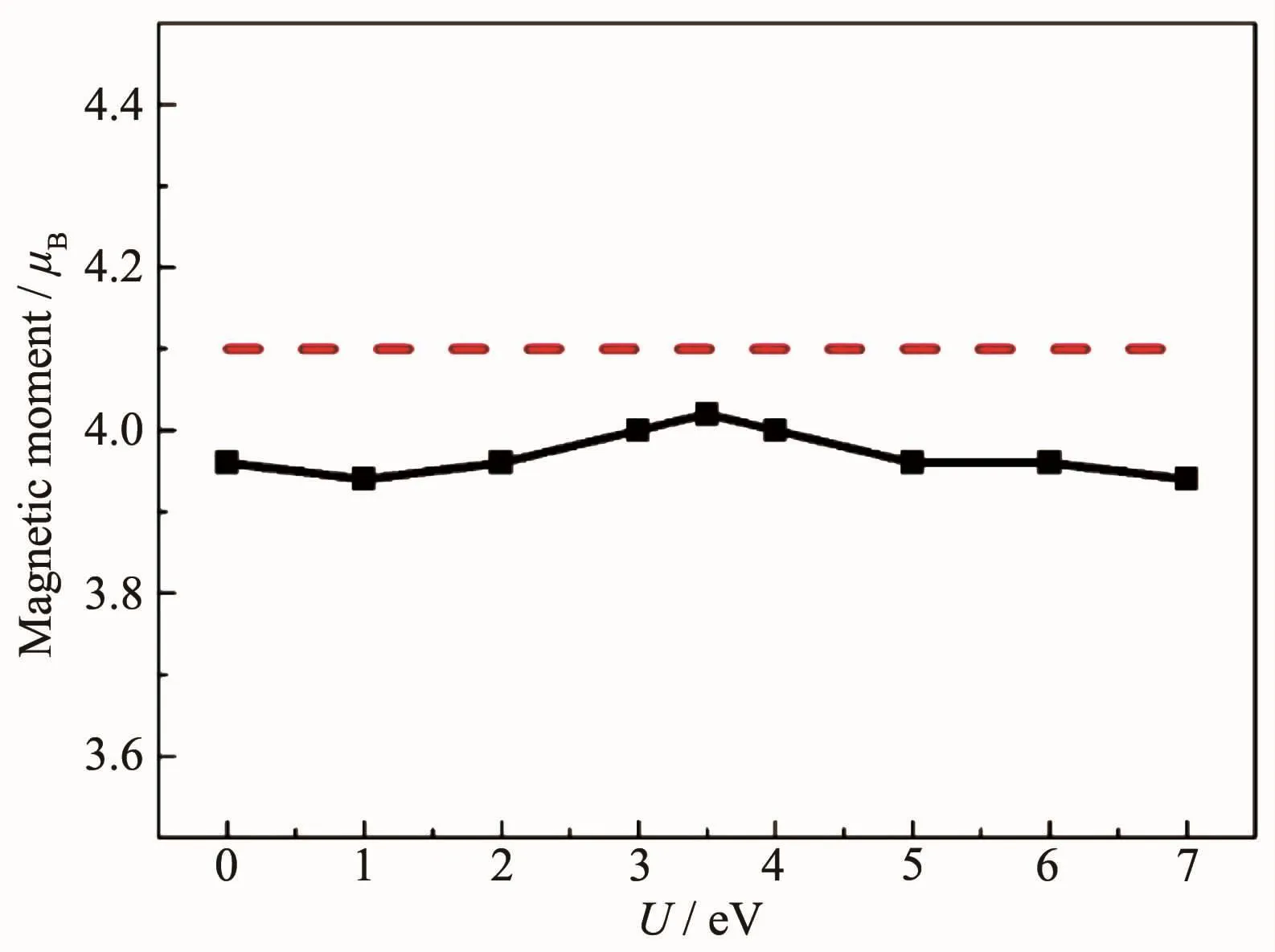
图4黑线:U分别取0,1,2,3,3.5,4,5,6,7 eV时,对应Fe3O4的磁矩;红线:实验测得Fe3O4的磁矩Fig.4Black:Magnetic moment of Fe3O4when U was set to 0,1,2,3,3.5,4,5,6 and 7 eV;Red: Experimental value
图5 是Hubbard U值取最优值3.5 eV时,计算得到的Fe3O4的态密度,可以看出,自旋向下的电子态密度越过费米能级,此方向的电子导电,表现金属性;而自旋向上的电子,费米能级处于能隙内,此方向的电子不导电。
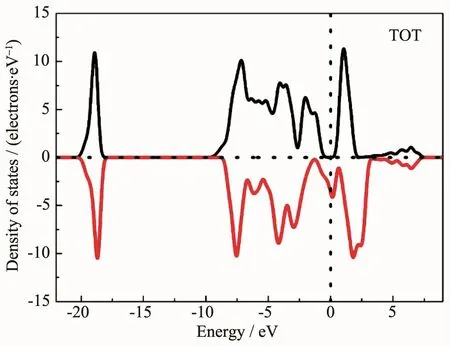
图5U取3.5 eV时,对应Fe3O4的态密度Fig.5Density of states of Fe3O4when U was set to 3.5
图6 是Fe3O4的分波态密度(PDOS),其中(a),(b)和(c)是Fe3O4的s,p和d轨道的分波态密度;(d),(e)和(f)是Fe3O4的A位Fe的s,p和d轨道的分波态密度;(g),(h)和(i)是B位Fe的s,p和d轨道的分波态密度;(j)和(k)是O的s和p轨道的分波态密度。其中涉及到的各原子电子结构为O2s22p4和Fe3s23p63d64s2,图(a)中,s轨道态密度的主峰呈现轴对称分布,几乎没有发生自旋极化并且和(d),(g),(j)图对比发现,主要来自O2s轨道;在费米面附近有极少的Fe4s电子,而Fe3s轨道能量与其他轨道的差别比较大,几乎不发生杂化而处于Fe离子内层。(b)图中p轨道发生微弱的自旋极化,和(e),(h),(k)图相比较发现p轨道态密度能量在-10~10 eV间的主峰主要来自O2p轨道,而能量在-20~-17.5 eV的小峰主要来自Fe3p轨道。(c)图中d轨道发生强烈的自旋极化,表明Fe3O4的分子磁矩主要来源于Fe的3d轨道自旋极化,并且对比(f)和(i)图发现FeA费米面以下自旋向下电子数比自旋向上电子数多,表明FeA磁矩为负;而FeB费米面以下自旋向上电子数比自旋向下电子数多,表明FeB磁矩为正,并且主要是B位Fe离子使Fe3O4表现半金属性。对比图(k),(f),(i)发现能量在-10~10 eV之间费米面附近,由于Fe和O的强烈共价作用使O2p和Fe3d存在杂化轨道。
表1是Fe3O4中各离子的详细计算结果,包括s,p,d和总的电子数,电荷数,离子磁矩,可以看出Fe3O4分子磁矩为4.02μB,其中A位Fe离子的磁矩是-3.94μB,B位Fe离子的磁矩是3.86μB,A位和B位Fe离子的磁矩相反,同时验证了Fe3O4为亚铁磁性半金属。
2.2 Fe3O4(001)表面结构和磁电性能
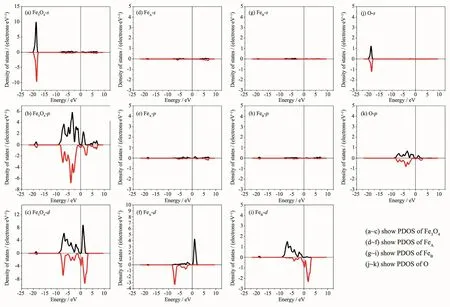
图6 Fe3O4体系分波态密度Fig.6PDOS of Fe3O4system
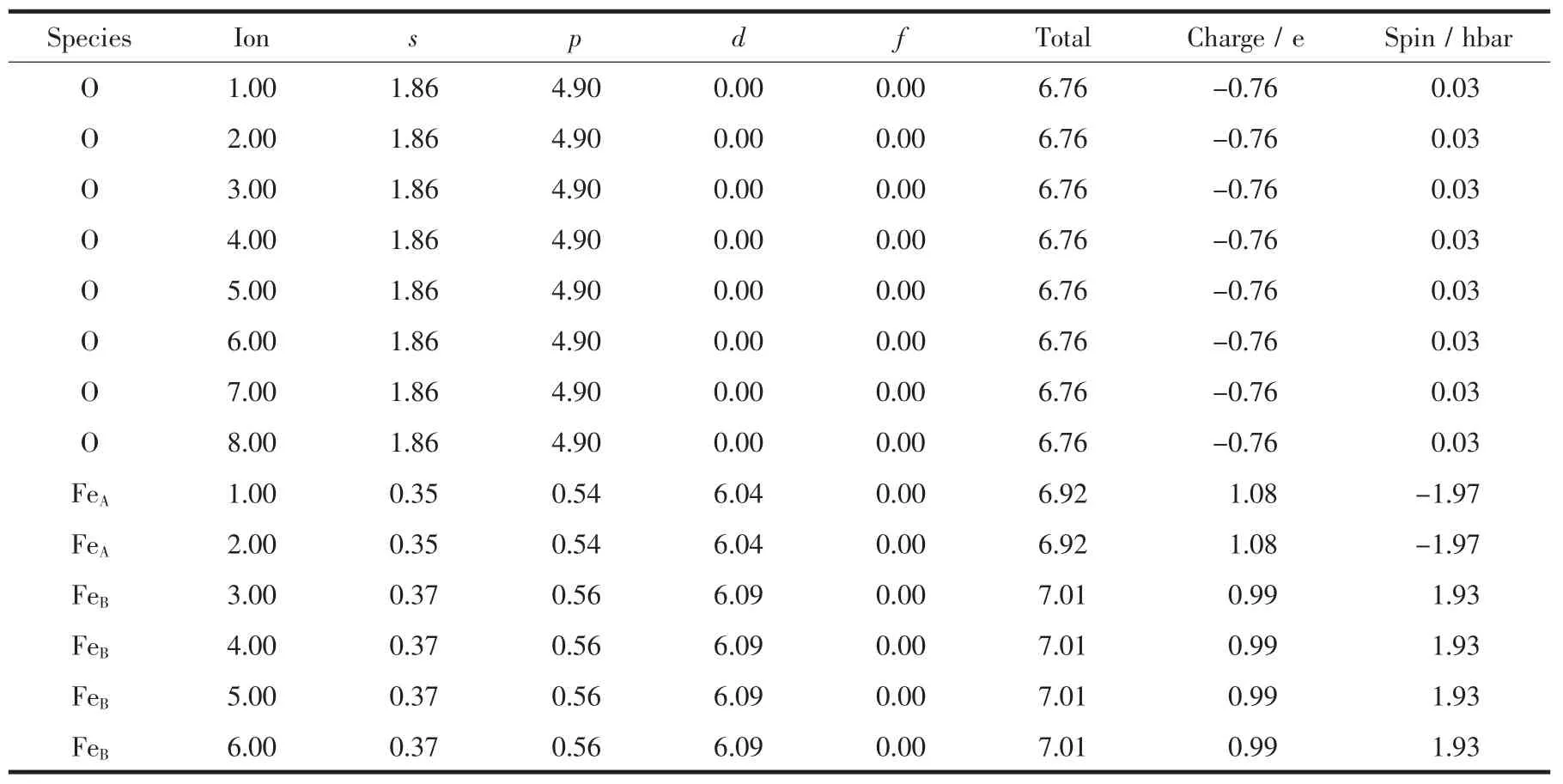
表1 Fe3O4中各离子参数:s,p,d和总电子数、电荷数、离子磁矩Table 1Electronic number of s,p,d and total orbits,charges and magnetic moments of ions
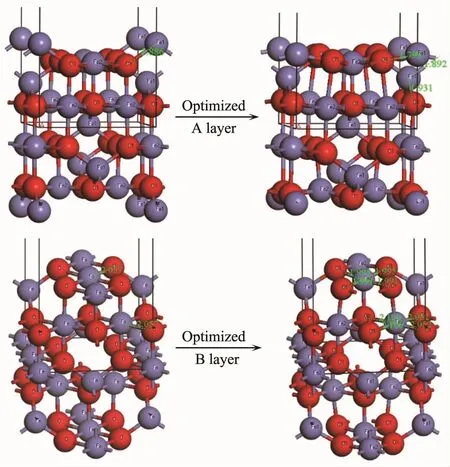
图7 Fe3O4(001)表面2个终端的表面结构Fig.7Two different terminations of Fe3O4(001)plane
Fe3O4的(001)表面是由A层和B层交替重叠堆积而成,其中A层终端包含四面体FeA,B层终端包含八面体FeB和O。图7是Fe3O4(001)表面2个终端的表面结构,A层终端优化后的第一层FeA-O键长是0.179 3 nm,比Fe3O4原胞中的0.188 6 nm短,表面第一层和第二层的层间距收缩了83%;B层终端优化后的FeB-O键长是0.199 1 nm和0.199 5 nm,比Fe3O4原胞中的0.205 7 nm短,表面第一层和第二层的层间距收缩了21.79%,表明2个终端都发生了表面变形。根据表面能的计算公式得到A层终端的表面能为1 179.70 eV·nm-2,B层终端的表面能为4 704.90 eV·nm-2,表明Fe3O4(001)表面的2个终端存在差异,由于A终端发生了强烈的表面变形,以至于表面A层与下方B层中的O十分接近,由于其电荷补偿的程度降低,因此其表面能也相对较低[9]。在实验上,A和B终端的获得依赖于处理条件,Tarrach等[28]通过实验研究得到A终端;Chambers等[29]在实验上观察到了带有缺陷的A终端;Wiesendanger等[30]通过实验在不同区域观察到A和B终端;Ceballos等[31]通过实验探究Fe3O4(001)表面A和B终端的存在形式,结果表明通过不同的制备过程可以得到不同的(001)表面终端,810 K下退火获得B终端,而相同的样品,通过多次溅射退火循环处理后,由于O2逐渐减少,最后得到A终端;Voogt等[32]则通过实验得到了B终端,并提出电荷补偿来自于O空位有序排列;Mariotto等[14]得到B终端并提出终端的获得依赖于八面体间隙Fe离子的电荷有序性。以上通过计算和实验都表明了A和B终端之间存在差异。
图8是Fe3O4(001)表面A层终端的态密度,可以看出A层终端表现半金属性,即自旋向下的电子态密度越过费米能级,而自旋向上的电子,费米能级处于能隙内。并且计算得到A层终端的总磁矩是11.92μB,其中各个FeA离子的磁矩范围为-3.74μB~-3.96μB,FeB离子的磁矩范围是3.78μB~3.86μB。
2.3 表面掺杂
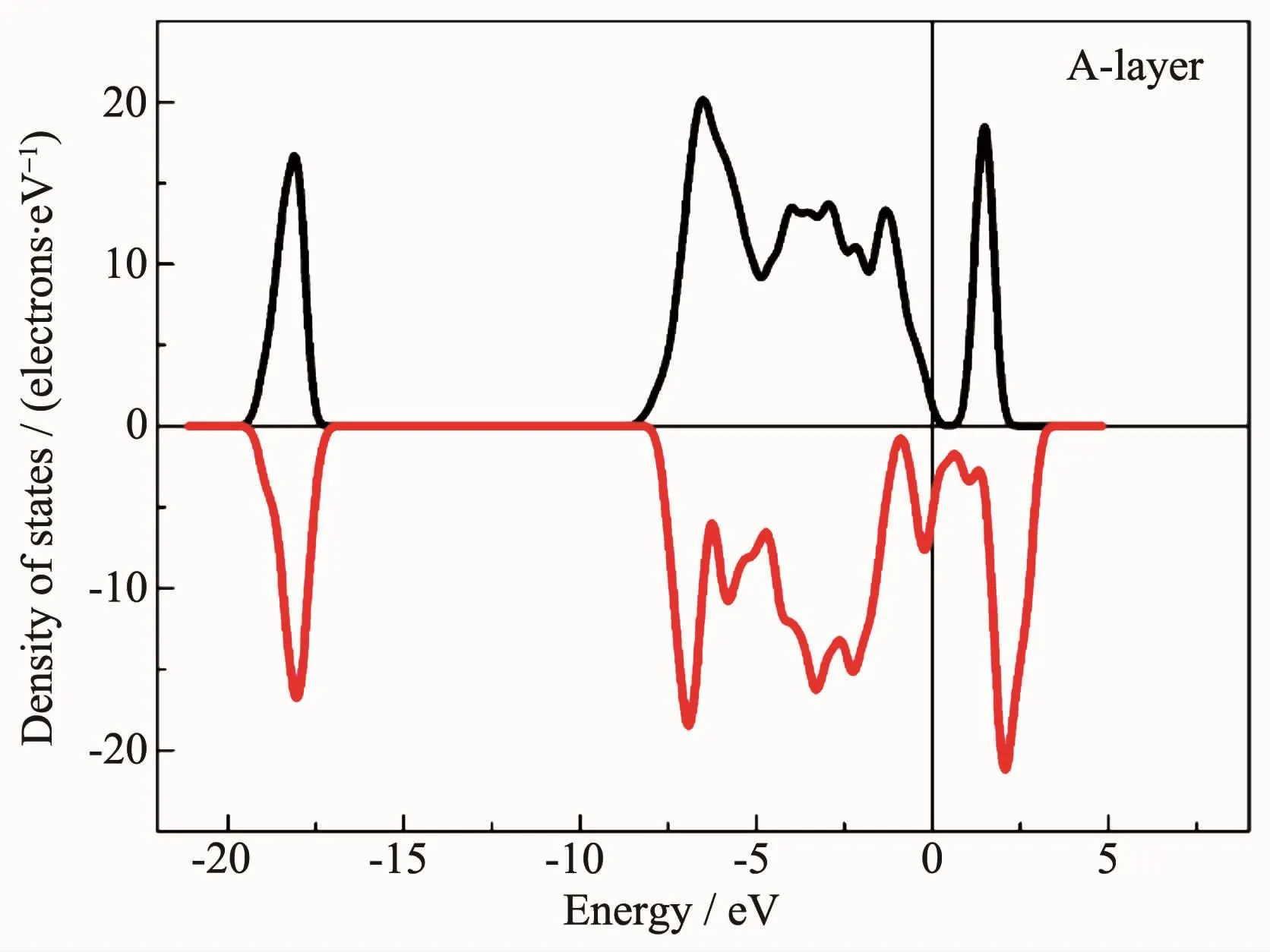
图8 Fe3O4(001)表面A终端的态密度Fig.8Density of states of Fe3O4(001)plane A-layer termination
分别用过渡元素V、Cr、Mn、Co、Cu和Zn取代Fe3O4(001)表面A层终端第一层的A位Fe形成新的表面,计算得到掺杂后的各个新表面仍然保持半金属性,即自旋向下的电子态密度越过费米能级而自旋向上的电子,费米能级处于能隙内。图9为掺杂后形成各个新表面的对应电荷密度图,表明掺杂离子和O之间都形成了强烈的共价相互作用[35-37]。分析计算结果得到V、Cr、Mn、Co、Cu和Zn取代A位Fe形成新表面的总磁矩分别为12.99μB,12.02μB,10.99μB,12.99μB,16.92μB和15.93μB,其中,对应掺杂离子的离子磁矩分别为-3.08μB,-4.25μB,-4.86μB,-2.52μB,0.07μB和0.03μB。可以看出Cr和Mn的离子磁矩都超过了被取代的(001)表面A终端第一层A位Fe离子的磁矩,Cr和Mn相比,Mn的磁矩更大,说明相同磁场中,Mn掺杂得到的新表面的传导电子能得到更大的自旋相关散射,从而产生较大的磁电阻效应[38-45]。计算得到V、Cr、Mn、Co、Cu和Zn取代A位Fe形成新表面的表面能分别为4 434.44、5 833.12、676.37、1 780.37、3 015.91和36 82.51 eV ·nm-2,可以得出稳定性从大到小依次为Mn、Co、Cu、Zn、V和Cr掺杂,并且Mn掺杂形成的新表面的表面能要比未掺杂的Fe3O4(001)表面A层终端的表面能1 179.70 eV·nm-2要小,因此Mn掺杂可以得到更稳定的表面。
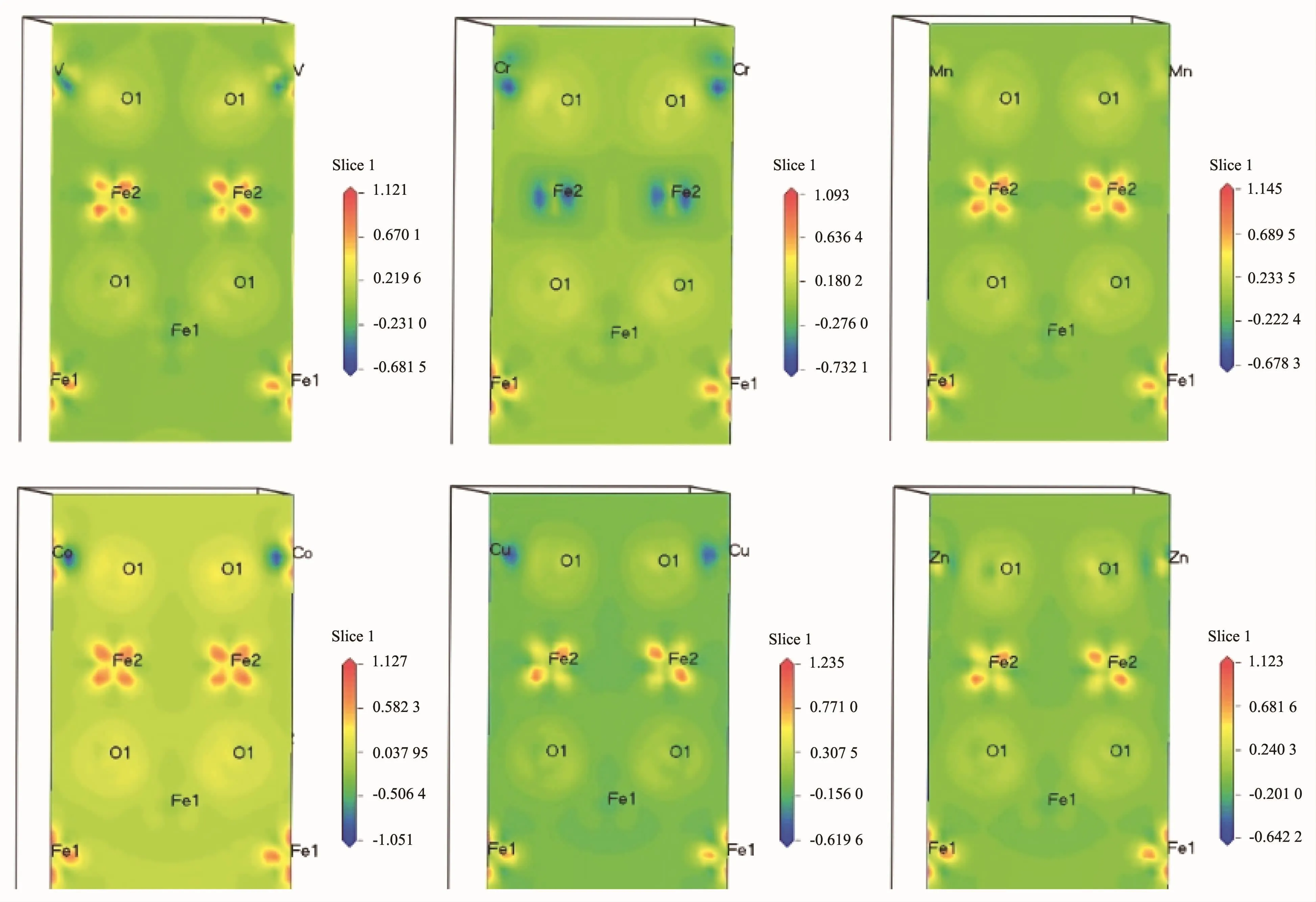
图9 Fe3O4(001)表面被V,Cr,Mn,Co,Cu和Zn掺杂后的电荷密度Fig.9Electron density of doped(001)plane A-layer termination
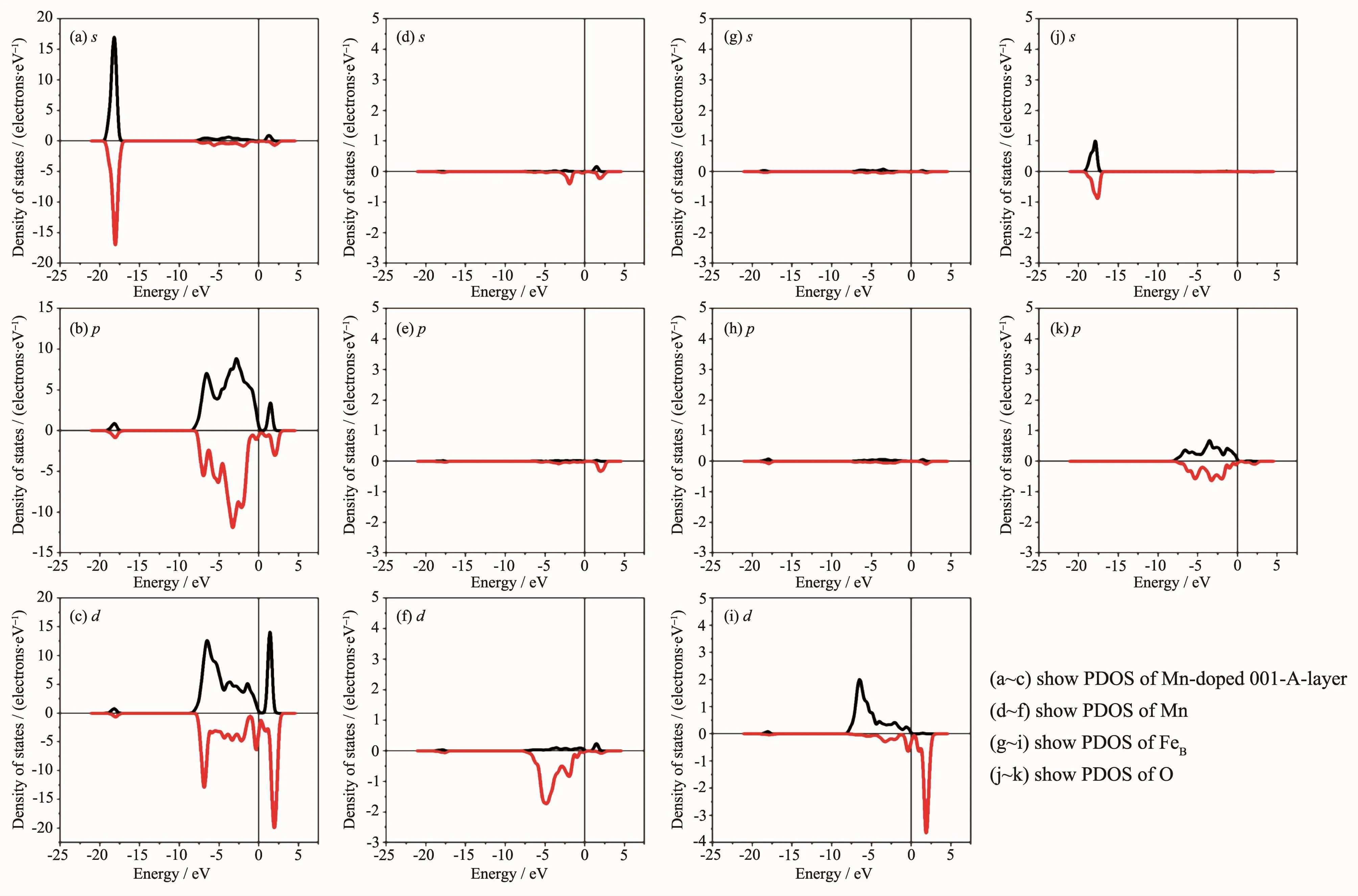
图10 Mn掺杂体系的分波态密度Fig.10PDOS of Mn-doped system
下面进一步分析Mn掺杂得到新表面的电子结构,图10是过渡元素Mn取代Fe3O4(001)表面A层终端第一层的A位Fe形成新表面的分波态密度(PDOS),其中(a),(b)和(c)是Mn掺杂Fe3O4(001)表面A层终端的s,p和d轨道的分波态密度;(d),(e)和(f)是Mn的s,p和d轨道的分波态密度;(g),(h)和(i)是B位Fe的s,p和d轨道的分波态密度;(j)和(k)是O的s和p轨道的分波态密度。其中涉及到的各原子电子结构为O2s22p4,Fe3s23p63d64s2和Mn3s23p63d54s2,图(a)和(d),(g),(j)图相比较,(a)图中能量在-20~-15 eV的峰主要来自O2s轨道,呈现轴对称分布,几乎没有发生自旋极化;并且在费米面附近有极少的Fe4s和Mn4s电子,对应的态密度比较平缓比重很小。(b)图中p轨道发生微弱的自旋极化,和(e),(h),(k)图相比较发现p轨道态密度能量在-10~5 eV间的主峰主要来自O2p轨道,而能量在-20~-17.5 eV的小峰主要来自Fe3p轨道。(c)图中d轨道发生强烈的自旋极化,表明总磁矩主要来源于Mn和Fe的3d轨道自旋极化,并且对比(f)和(i)图发现Mn3d电子发生了强烈的自旋极化,并且费米面以下自旋向下电子数比自旋向上电子数多,表明Mn磁矩为负;而FeB费米面以下自旋向上电子数比自旋向下电子数多,表明FeB磁矩为正,并且主要是B位Fe离子使新表面表现半金属性。对比图(k),(f),(i)发现能量在-10~5 eV之间费米面附近,Mn3d,Fe3d和O2p存在杂化轨道。
3 结论
本文运用第一性原理计算方法,基于密度泛函理论,对Fe3O4,Fe3O4(001)表面及过渡元素掺杂表面形成新表面的表面结构,稳定性,电子结构,分子磁矩等进行了计算和分析。结果表明:在GGA+U计算中Hubbard U值取3.5 eV时,得到亚铁磁性半金属Fe3O4的磁矩与实验值最接近。比较Fe3O4(001)表面的2个不同终端的表面能得到A终端较稳定。用过渡金属元素V、Cr、Mn、Co、Cu和Zn对Fe3O4(001)表面A终端第一层的FeA进行替换,得到的新表面结构中Mn掺杂表面最稳定,同时Mn的3d电子发生了非常强烈的自旋极化劈裂,使磁矩明显增大。该掺杂材料的传导电子将获得更大的自旋相关散射,从而产生更大的磁电阻效应,研究在核自旋共振压力显微、自旋学和自旋电子学中具有重要价值。
[1]Degroot R A,Mueller F M,Vanengen P G,et al.Phys.Rev. Lett.,1983,50(25):2024-2027
[2]Watts S M,Wirth S,von Molnar S,et al.Phys.Rev.B,2000, 61(14):9621-9628
[3]Park J H,Vescovo E,Kim H J,et al.Nature,1998,392(6678): 794-796
[4]REN Shang-Kun(任尚坤),ZHANG Feng-Ming(张凤鸣),DU You-Wei(都有为).Prog.Phys.(物理学进展),2005,24:381-397
[5]Coey J,Viret M,von Molnar S.Adv.Phys.,1999,48(2):167-293
[6]Pickett W E,Moodera J.Phys.Today,2001,54(6):83
[7]LIU Jun(刘俊).Thesis for the Doctorate of Chongqing University(重庆大学博士论文).2008.
[8]Pentcheva R,Wendler F,Meyerheim H L,et al.Phys.Rev. Lett.,2005,94(12):126101
[9]Cheng C.Phys.Rev.B,2005,71(5):52401
[10]Szotek Z,Temmerman W M,Svane A,et al.Phys.Rev.B, 2003,68(5):054415
[11]Szotek Z,Temmerman W M,Svane A,et al.J.Phys.:Condens. Matter,2004,16(48):S5587
[12]Spiridis N,Barbasz J,Lodziana Z,et al.Phys.Rev.B,2006, 74(15):155423
[13]Spiridis N,Handke B,Slezak T,et al.J.Phys.Chem.B,2004, 108(38):14356-14361
[14]Mariotto G,Murphy S,Shvets I V.Phys.Rev.B,2002,66 (24):245426
[15]Yu X,Huo C,Li Y,et al.Surf.Sci,2012,606(9):872-879
[16]WANG Hong-Ming(王宏明),ZHENG Rui(郑瑞),LI Gui-Rong(李桂荣),et al.Chinese J.Inorg.Chem.(无机化学学报),2015,31(11):2143-2151
[17]Segall M D,Lindan P,Probert M J,et al.J.Phys.:Condens. Matter,2002,14(11):2717-2744
[18]WU Guo-Hao(吴国浩),ZHENG Shu-Kai(郑树凯),LÜ Xiao (吕霄).Chinese J.Inorg.Chem.(无机化学学报),2013,29(1): 9-14
[19]Monkhorst H J,Pack J D.Phys.Rev.B,1976,13(12):5188-5192
[20]Vanderbilt D.Phys.Rev.B,1990,41(11):7892-7895
[21]Kresse G,Hafner J.J.Phys.:Condens.Matter,1994,6(40): 8245-8257
[22]Weinert M,Wimmer E,Freeman A J.Phys.Rev.B,1982, 26(8):4571-4578
[23]Dudarev S L,Botton G A,Savrasov S Y,et al.Phys.Rev.B, 1998,57(3):1505-1509
[24]Steynberg P J,van den Berg J A,van Rensburg W J.J.Phys.: Condens.Matter,2008,20(6):64238
[25]Jordan K,Cazacu A,Manai G,et al.Phys.Rev.B,2006,74 (8):85416
[26]Mulakaluri N,Pentcheva R,Scheffler M.J.Phys.Chem.C, 2010,114(25):11148-11156
[27]Mulakaluri N,Pentcheva R,Wieland M,et al.Phys.Rev.B, 2009,103(17):176102
[28]Tarrach G,Burgler D,Schaub T,et al.Surf.Sci.,1993,285 (1):1-14
[29]Chambers S A,Thevuthasan S,Joyce S A.Surf.Sci.,2000,450(1):273-279
[30]Wiesendanger R,Shvets I,Burgler D,et al.Science,1992, 255(5044):583-586
[31]Ceballos S F,Mariotto G,Jordan K,et al.Surf.Sci.,2004, 548(1):106-116
[32]Voogt F,Fujii T,Smulders P.Phys.Rev.B,1999,60(15): 11193
[33]Zhou Z L,Wang Y L,Wu Jonathan Q M,et al.IEEE Trans. Inf.Forensic.Secur.,2017,12(1):48-63
[34]Xia Z H,Wang X H,Zhang L G,et al.IEEE Trans.Inf. Forensic.Secur.,2016,11(11):2594-2608
[35]Ren R,Ren Y J,Li X.J.Alloys Comp.,2016,662:290-295
[36]Ren R,Ren Y J,Li X.Prog.Nat.Sci.:Mater.Int.,2016,26(2): 173-176
[37]Ren R,Ren Y J,Wang W R,et al.Sci.Adv.Mater.,2015,6 (6):1255-1261
[38]Ren R,Li X,Wang W R,et al.J.Appl.Phys.,2013,114 (13):133705
[39]Ren R,Wang W R,Li X,et al.Phys.Scr.,2014,89(3): 035601
[40]Ren R,Ren Y J,Li X,et al.Optoelectron.Adv.Mater. Rapid Commun.,2015,9(11/12):1453-1457
[41]REN Ren(任韧),XU Jin(徐进),REN Da-Nan(任大男).Acta Phys.Sin.(物理学报),2010,59(11):8155-8159
[42]REN Ren(任韧),XU Jin(徐进),ZHU Shi-Hua(朱世华). Acta Phys.Sin.(物理学报),2006,55(2):555-563
[43]Ren R,Ren Y J,Wu Z,et al.Optoelectron.Adv.Mater. Rapid Commun.,2015,9(7/8):956-960
[44]Zhao Z X,Ren R,Li X,et al.J.Nanosci.Nanotechnol., 2017,17(3):1957-1962
[45]Xia Z H,Wang X H,Sun X M,et al.IEEE Trans.Parallel Distrib.Syst.,2016,27(2):340-352
Theoretical Study of the Magnetic and Electric Properties of Transition Elements Doped Fe3O4(001)Surface
ZHAO Zhong-XiaREN Ren*REN Yi-JingZHOU Zhi-Li
(School of Science,Xi′an Jiaotong University,Xi′an 710049,China)
The electronic structures and magnetic properties of Fe3O4,Fe3O4(001)surface and transition elements -doped(001)surfaces were calculated based on the density functional theories.The results show that the halfmetallicity of Fe3O4is from Fe-ions on B-sites,and magnetic moment is from spin polarization of 3d-orbits.For the Fe3O4(001)surface,the A-layer termination is more stabilized with half-metal character.Fe-ions on A-sites of A-layer termination are substituted by doped V-ions,Cr-ions,Mn-ions,Co-ions,Cu-ions and Zn-ions so that the new surface structures are designed.All of them have half-metal character,and the Mn-doped surface is the most stabilized with greater magnetic moment.
Fe3O4;density functional theory;electronic structures;magnetic property;surface energy
O471.4;O614.81;O743+3
A
1001-4861(2017)06-0923-09
10.11862/CJIC.2017.091
2016-03-09。收修改稿日期:2017-03-21。
国家自然科学基金(No.61574115,61681240392,10775111)和陕西省自然科学基金项目(No.2016JM1029),PAPD,CICAEET资助。
*通信联系人。E-mail:renr01@163.com,renrray@163.com
猜你喜欢
杂志排行

无机化学学报的其它文章
- CoAl2O4/蜂窝陶瓷催化剂的制备及其催化臭氧化性能
- Photocatalytic Hydrogen Production Based on Cobalt-Thiosemicarbazone Complex with the Xanthene Dye Moiety
- 两种金属-有机钙钛矿材料的负热膨胀性质
- Pyrazolate-Based Dipalladium(Ⅱ,Ⅱ)Complexes:Syntheses,Characterization and Catalytical Performance in Suzuki-Coupling Reaction
- 以滤纸为模板合成新型介孔生物活性玻璃微管材料
- Br-掺杂Bi2WO6的水热法合成及其可见光催化性能