美国和欧盟的罕用药研发激励政策对比研究与启示Δ
2017-07-03田丽娟沈阳药科大学工商管理学院沈阳110016
杨 莉,田丽娟,林 琳(沈阳药科大学工商管理学院,沈阳 110016)
·药事管理·
美国和欧盟的罕用药研发激励政策对比研究与启示Δ
杨 莉*,田丽娟,林 琳(沈阳药科大学工商管理学院,沈阳 110016)
目的:为构建和完善我国的罕用药研发激励政策提供参考和建议。方法:从罕用药的立法沿革、研发罕用药的激励措施与效果方面对美国和欧盟的罕用药研发激励政策进行对比,并为我国完善相关政策提供建议。结果与结论:美国与欧盟的罕用药激励政策分别始于1983年美国《罕用药法案》与1999年欧盟《罕用药管理规范》,之后通过不断完善,形成了较为完备的体系。美国与欧盟在罕用药的认定标准、认定程序、具体激励措施(研发资助、税收减免、费用减免、微型与中小企业额外激励、市场独占、特殊审批程序)等方面有所差异,如在费用减免方面,美国对处方申请费用、生产费用和药物确认费用进行减免,而欧盟对协议帮助费用、初始和后续要求费用,审批前的检查费用和首次上市申请费用依类型按一定比例进行减免。罕用药激励政策推行后,其资格认定数量及上市数量大幅增加、微型与中小型企业成为罕用药研发的生力军、研发投资涵盖各类疾病治疗领域、罕用药研发成为药物创新和生物技术发展的主要方向。我国应该尽快确定罕用药研发激励的相关立法、设立罕用药的资格认定、从多方面入手完善罕用药研发激励具体措施,同时加强与其他国家在罕用药资格认定和研发激励方面的合作。
罕用药;研发激励;美国;欧盟;对比;研究
罕用药由于临床研究开展困难、研发成本难以收回等因素成为许多医药企业不愿意涉足的领域。目前,世界上超过35个国家都推行并实施了不同程度的罕用药研发激励政策[1]。美国和欧盟作为世界上对罕用药研发激励最为倾斜的两个国家(地区),是许多学者研究的对象,但是针对这两个国家(地区)的罕用药研发激励政策的对比研究较少。因此,在本研究中,笔者从罕用药的认定、立法、具体措施以及实施效果等多方面对美国和欧盟的罕用药研发激励政策进行对比研究,以期为我国罕用药研发激励政策的构建和完善提供建议。
1 美国和欧盟罕用药研发激励的立法沿革
1983年1月4日,美国颁布并实施了《罕用药法案》,开启了通过立法对罕用药进行研发激励之门。1999年,欧盟委员会颁布了《罕用药管理法规》,鼓励欧盟的医药企业进行罕用药的研发,该法于2000年4月28日正式实施。美国和欧盟罕用药研发激励立法比较见图1。

图1 美国和欧盟罕用药研发激励立法比较Fig 1 Comparisons of the R&D incentive legislations for orphan drugs in USA and the EU
由图1可知,美国对罕用药资格认定的范围不断放宽,研发激励措施也逐渐加强。1992年和1994年曾有两次限制罕用药市场独占权的修订提案,而国会最终基于“药虽贵但强于无药可医”的保守考虑未通过此项修订案,这也反映了美国对罕用药研发激励的强烈倾斜态度[2]。除了《罕用药法案》之外,美国还于1992年颁布了《罕用药法实施办法》,对罕用药的研发激励和管理进行了更为详细的规定;2002年通过了《罕见疾病药物发展法案2002》,对《罕用药法案》的内容进行了补充。
欧盟继《罕用药管理法规》之后多次颁布新的法规继续补充和完善罕用药研发激励的内容。相比于美国,欧盟对罕用药资格的限定更多。此外,欧盟药品管理局(EMA)还出台了一系列指南,对罕用药研发享受到的一系列优惠政策,如费用减免、协议帮助等加以说明。
2 美国和欧盟罕用药研发激励的具体措施比较
2.1 罕用药资格的认定标准
美国罕用药资格的获得需要满足以下条件:治疗疾病的患病人数不满20万,或者患病人数超过20万,但合理预期的销售收入无法收回研发成本。2013年8月实施的《罕用药法案》修正案,将常见疾病的罕见临床类型的治疗药物也纳入可获得罕用药资格的范围。
欧盟罕用药资格只授予用来预防、诊断和治疗危及生命、慢性消耗性疾病的药品,并且该疾病在欧洲患病人数不超过万分之五,或者如果没有激励措施,该药品上市后的收入无法充分地回收其研发成本。除此之外,还应该满足以下条件:除了该药品,目前还没有令人满意的预防、诊断和治疗该疾病的药品或方法,或者相比于现存的药品或方法,该药品对患者有显著收益(具有临床优势或对患者有显著的贡献,如更好的治疗指征、不同的作用机制、更高的安全性、更方便的给药途径以及更便宜的价格等)[3]。
典型国家或地区的罕用药资格认定标准见表1。
由表1可见,美国执行了最宽松的标准,而欧盟则执行了最严格的标准。

表1 典型国家或地区的罕用药资格认定标准Tab 1 Orphan drug designation standards in typical countriesor regions
2.2 罕用药资格的认定程序
美国罕用药资格的认定部门是FDA的罕用药开发办公室(OOPD)。欧盟的罕用药资格由EMA设立的罕用药委员会(COMP)负责审评,并出具评估意见,但由欧盟委员会(EC)最终决定是否授予罕用药资格。美国和欧盟的罕用药资格认定程序见图2。

图2 美国和欧盟的罕用药资格认定程序Fig 2 O rphan d rug designation procedure in USA and the EU
由图2可见,欧盟对罕用药资格的认定程序更为复杂。除此之外,在审评的具体细节上,美国和欧盟也有较多区别,详见表2。

表2 美国和欧盟的罕用药资格认定具体内容比较Tab 2 Comparison of the specific contents of orphan drug designation in USA and the EU
由于欧盟在罕用药的认定上有严格的时限要求,因此从提交正式申请算起,整个罕用药资格的认定为120~180天。而美国对时限无明确要求,因此整个罕用药的认定为30~365天,其中有75%的认定都是在90天内完成的。随着近年来申请数目的增加,FDA罕用药认定时限的目标是将75%的申请控制在120天内完成审核。截至2015年12月31日,FDA共收到5 561份申请,其中有3 612份申请获得了罕用药资格认定,通过率为65%[4];欧盟共收到2 385份申请,得到罕用药资格的申请为1 596份,通过率为67%[5]。因此,从认定的效率和通过率来说美国和欧盟的差别不大。虽然美国在提交申请前没有专门的协助会议,但是在申请过程中FDA也会给申请人提供一些建议。
2.3 激励措施
美国和欧盟的罕用药研发激励措施比较见表3。

表3 美国和欧盟的罕用药研发激励措施比较Tab 3 Com parison of the orphan drug incentivemeasures in USA and the EU
2.3.1 研发资助 美国的罕用药研发资助:一是美国国立卫生研究院(NIH)提供的项目资助,倾向于罕用药研发的基础研究;二是一些公司合作伙伴组织(PPPs)提供的针对一些特定疾病的研究的资助,例如国际艾滋病疫苗倡议组织(IAVI);三是OOPD办公室的罕用药研发项目,主要资助的是罕用药的临床研究,该项目1983年起实施以来,迄今已经收到1 800多份申请,有600余项项目得到了资助,资助的项目中已经有45个罕用药获得了上市批准[6]。
EMA不直接对罕用药的研发提供资助,由EC通过框架计划对罕用药的研发提供资金支持。《第七框架计划(2007-2013)》(FP7)从2009年起将获得罕用药资格作为获得资助的必要条件。FP7共投资6.2亿欧元资助超过120个罕用药研发项目,资助范围包括罕用药的基础研究、临床前研究和临床研究,涵盖各类罕见疾病。2014年,新的研究与创新框架计划——“地平线2020”(Horizon 2020)正式启动,为期7年(2014-2020年)。2014-2015年,“地平线2020”已经资助了肿瘤、呼吸、消化等方面共20余个罕用药研发。
2.3.2 税收减免 美国的罕用药税收减免用于临床试验阶段的支出,无论该罕用药最终能否获得上市许可都可享受减免。美国国会的技术评估办公室(OTA)的研究表明,税收减免可以将临床研究成本至少降低24%,仅在Ⅲ期临床阶段,罕用药的研发成本就是非罕用药的1/2[7]。由于临床试验是整个药物研发过程中耗资最大的阶段,因此税收减免带来的收益非常可观,也成为很多医药企业是否继续开展临床研究的重要考量因素。
考虑到享受税收减免的罕用药最终不一定能够获得上市,资金不一定能得到最佳使用,故欧盟并未制定税收减免政策,而是通过设立更长的市场独占期来代替税收减免。
2.3.3 费用减免 美国的罕用药费用减免分为处方申请费用、生产费用和药物确认费用。如果获得罕用药资格,处方药申请费是自动豁免的。生产费用和药物确认费的减免则需要提交申请,减免的比例不固定。
欧盟的费用减免需要提出申请,包括协议帮助费用、初始和后续要求费用,审批前的检查费用和首次上市申请费用,减免的比例是固定的。
2006-2015年美国和欧盟的罕用药的费用减免总额均呈现逐年上涨的趋势,详见表4(数据来源于医药市场研究机构Evaluate Pharma)。

表4 2006-2015年美国和欧盟罕用药费用减免总额Tab 4 Com parison of fee reduction of orphan drugs in USA and the EU during 2006-2015
2.3.4 协议帮助 美国和欧盟的协议帮助主要包括罕用药资格的保持和罕用药的批准上市。协议帮助的方式是由申请人向OOPD或人用药品委员会(CHMP)提出申请,并列出相关问题,由OOPD和CHMP给予解答以解决罕用药在研发过程中有关问题,降低因研究设计不当而导致的取消罕用药资格和未通过上市批准的风险。申请人可以申请协议帮助的次数没有限制。协议帮助在帮助罕用药上市方面发挥了积极的作用,截至2015年底,CHMP共完成了951份协议帮助,被批准上市的罕用药中,超过50%都接受过协议帮助。
2.3.5 SEMs额外激励 欧盟从2005年(罕用药的申请必须通过集中审批程序批准)后开始对SEM s施行额外的罕用药研发激励政策。企业需要向EMA提交申请,由EMA审核确定其满足SEMs的标准,才能享受此激励。截至2015年底,欧盟已批准上市的罕用药中,有25个来自SEMs。
2.3.6 市场独占 美国和欧盟分别给予罕用药7年、10年的市场独占期。如果该罕用适应证可以用于儿科,并进行了儿科临床研究,则独占期可以分别延长6个月、2年。同时,美国和欧盟也规定了丧失罕用药资格的情况:某药品比已上市的药品更具有临床优越性(更有效、更安全或对患者的健康更有益);或获得罕用药独占的药品供应不足,无法满足需求;或获得罕用药独占权的药品持有人同意。
此外,欧盟还规定,当罕用药5年独占期结束,如果有成员国通知EMA该药品已经不符合罕用药资格的标准,则该罕用药的市场独占期将会被降为6年。
2.3.7 特殊审批 美国和欧盟的罕用药特殊审批程序比较见表5。

表5 美国和欧盟的罕用药特殊审批程序比较Tab 5 Com parison of special approval procedure of orphan drugs in USA and the EU
从表5可以看出,美国的加速审评和优先审评与欧盟的条件审评和加速审评类似,而其他的审评程序美国和欧盟各有特色。申请人想要通过特殊审评程序获得上市批准必须向FDA或EMA提出申请。截至2015年9月底,FDA批准罕用药所需的平均时间为315天,非罕用药为378天;EMA批准罕用药所需的平均时间为210天,非罕用药为270天[8]。可见,罕用药上市速度明显要快于非罕用药。
2.4 美国和欧盟在罕用药研发激励方面的合作
2.4.1 罕用药资格申请 2007年,FDA和EMA合作引进了罕用药资格通用申请格式,激励申请人在美国和欧盟并行提交申请。通用格式既包含了FDA和EMA的共同信息要求,也包含各机构的单独要求,申请人可以以通用格式同时在美国和欧盟提出申请。通用申请格式的引进大大降低了申请人的行政成本,提高了申请效率。迄今为止,EMA的罕用药资格申请有50%左右是并行提交到FDA的,有40%左右使用了通用申请格式。
2.4.2 年度报告 获得罕用药资格后,FDA要求14个月内提交年度发展报告,此后每年提交1次直至获得上市批准。EMA也有提交年度发展报告的要求。2011年开始,美国和欧盟宣布接收通用模板的年度报告。年度报告的内容主要包括研究计划、已完成的研究工作、遇到的困难以及潜在的风险等。依据研究报告的内容,FDA和EMA有权撤销罕用药资格。
2.4.3 协议帮助 FDA和EMA建立了并行的协议帮助程序。当申请人获得罕用药资格后,可以同时向FDA和EMA申请协议帮助。同时FDA和EMA也可通过协商,对申请人同时开展协议帮助。
FDA和EMA的合作,也促进了很多医药企业积极申请在美国和欧盟同时获得认定和上市。截止到2015年底,欧盟获得罕用药资格的药品中有53%同时在美国获得了罕用药资格,获得上市的罕用药中有49%同时在美国获得了上市[9]。
3 美国和欧盟的罕用药研发激励效果比较
推出罕用药激励政策之后,美国和欧盟的罕用药资格认定情况和批准上市情况见图3(数据来源于FDA和EMA网站[4-5]),效果比较见表6(数据来源于Drug Discovery Today,其中V类别是解剖学治疗学及化学分类系统代码中的规定)。
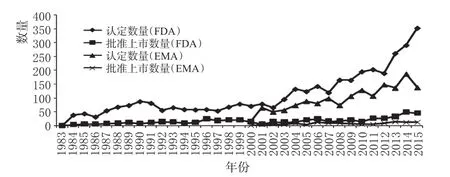
图3 美国和欧盟的罕用药资格认定和批准上市情况Fig 3 Orphan drug designation and approval for listing in USA and the EU
3.1 罕用药资格认定数量及上市数量大幅增加
从图3可以看出,美国和欧盟的罕用药资格的认定数量均呈现逐年上涨趋势,且每年都有一定数量的罕用药获得上市,上市的罕用药总量呈现平稳增长。
从取得上市资格的罕用药占获得罕用药资格的药品的比例来看,美国为15.1%,欧盟为7.6%。虽然欧盟的罕用药激励政策比美国滞后17年左右,但是资格认定的增长速度很快。但是资格认定中获得上市批准的比例却比美国低很多。如果以罕用药上市的数量和比例作为衡量激励政策作用的重要标准,则美国的激励政策效果更为显著。
3.2 中小型企业成为罕用药研发的生力军
由表6可见,在申请类型上美国和欧盟极为相似。提出罕用药资格申请最多的公司集中在全球排名100以后的制药公司。一些中小企业,特别是一些生物科技公司看准了罕用药研发激励政策(特别是市场独占政策)提供的资金支持和研发帮助,以罕用药作为目标市场,在此基础上逐渐发展壮大,比如美国的BioMarin、Ae-gerion制药公司,欧盟的Actelion制药公司。

表6 美国和欧盟的罕用药激励政策的效果比较Tab 6 Comparison of incentive policy effects of orphan d rugs in USA and the EU
3.3 罕用药研发投资涵盖各类疾病领域
由表6可见,美国和欧盟申请数量排第1位的都是在研发投资上更容易回收的抗肿瘤和免疫调节药物。同时,在其他治疗领域上,美国和欧盟的罕用药研发资本均有涉足。且在治疗领域分类的申请排序上,美国和欧盟的差别也不大。
3.4 罕用药研发已经成为药物创新和生物技术发展的主要方向
由表6可见,美国和欧盟的药物类型,排名第1位的均为小分子制剂,第2位为生物制剂。近年来,随着研发突破越来越困难,研发投资越来越多,很多制药公司将研发战略从常见病药物转移到罕用药的研发上,生物医药产业成为罕用药研发的主力。因此,美国近些年来批准上市的罕用药物占每年新化学药物的1/3左右,占每年新生物药物的2/3左右[10]。
4 美国和欧盟罕用药研发激励政策对我国的启示
我国目前尚未建立类似于美国和欧盟的系统的罕用药研发激励政策。主要的有关罕用药研发激励的政策包括:《药品注册管理办法》第45条规定对罕见病药物可以进行特殊审批;《新药注册特殊审批管理规定》中提出,在必要情况下,罕见病药物可以提出减少临床受试人群或豁免临床试验的申请;《关于药品注册审评审批若干政策的公告》中提出对罕见病等疾病的创新药注册申请实行单独排队,加快审评审批。因此,目前我国罕用药的研发激励政策主要集中在特殊审评,与美国和欧盟的相关政策存在较大差距。
4.1 确定罕用药研发激励的相关立法
美国和欧盟罕用药研发激励的立法层级较高,都属于立法委员会通过的法律。我国目前最高层级的与药品相关的专门立法是《中华人民共和国药品管理法》,但无专门的罕用药研发激励相关条款。涉及到罕用药研发激励的上述条款属于部门规章或补充和说明,条款比较简单,缺乏详细具体的内容。因此,我国首先需要解决的是罕用药研发激励的立法问题。笔者建议我国在《药品管理法》中增加药品研发激励的相关条款,例如鼓励和促进罕用药的研发,并由国家食品药品监督管理总局出台详细的政策解释,完善《药品注册管理办法》中关于罕用药注册的相关条款。
4.2 设立罕用药的资格认定
目前,我国的相关法规和文件中出现的都是“罕见病药物”的表述,强调的是药物所治疗疾病的患病率,但是即使对罕见病也没有作确切定义。因此,我国罕用药的资格认定首先应该明确罕见疾病的定义。在罕用药资格判定方面,除日本以外,其他国家都将无法收回投资作为可以获得罕用药资格的条件之一。很多创新药物都会面临上市后多年无法收回成本的尴尬境地,因此我国可以考虑将这类药物纳入罕用药的范围;同时,应借鉴欧盟设立限制条件,例如加入针对一定危重程度的疾病、具有临床优越性等要求。此外,研究项目的可行性通过申请研发资助时加以审评,笔者认为并非必要条件。是否被其他管理当局拒绝授予罕用药资格仅作为参考条件,因为每个国家和地区的罕见疾病并不相同。
笔者建议我国成立专门的罕用药资格认定机构,对罕用药资格进行审评;确定罕用药资格的认定程序,并将罕用药资格认定程序作为罕用药注册批准的前置程序;由于我国目前的药品行政审批都有具体的时限要求,因此对罕用药资格认定也要设立具体的时限;此外,相关部门应做好信息公开工作,及时在相关网站上对获得罕用药资格的药品进行公开,并接受社会监督;年度发展报告应该作为维持或撤销罕用药资格的必要条件。
4.3 完善罕用药研发激励具体措施
4.3.1 研发资助 我国每年投入大量的资金对新药研发进行资助,例如重大新药创制、国家重点新产品计划等。对罕用药研发提供资金支持是研发激励最直接的措施,建议我国设立罕用药研发的专项资助,做到临床与基础研究并重。相关部门可以针对特定疾病设立专项,引导资本投入到市场不愿涉足的疾病领域中去。此外,获得研发资助也需要经过申请和筛选,使资金切实运用到具有可行性的项目上。
4.3.2 费用减免 相比于美国和欧盟,我国目前还未推行处方药申报付费制度,因此费用减免在当前意义不大,但是可以免除协议帮助费用。
4.3.3 税收减免 关于税收减免,笔者建议采用欧盟的形式,可以不制定。第一由于税收减免不是靠国家食品药品监督管理总局一个部门就可以决定的,这涉及到我国的税务部门,会增加政策制定的难度;第二,获得罕用药资格的药品不一定能够获得上市,如果没有获得上市,税收减免的后果则是由政府来承担的。
4.3.4 协议帮助 我国《药品注册特殊审批管理规定》中明确提出,已获准实行特殊审批的注册申请,原国家食品药品监督管理局药品审评中心应建立与申请人沟通交流的工作机制,共同讨论相关技术问题;申请人在完成某一阶段临床试验及总结评估后,可就下列问题向原国家食品药品监督管理局药品审评中心提出沟通交流申请:重大安全性问题、临床试验方案和阶段性临床试验结果的总结与评价。这其实已经是协议帮助的一部分了。我国可以借鉴美国和欧盟的经验,在获得罕用药资格前,或者提交注册前就建立起与国家食品药品监督管理总局的沟通,对罕用药资格的认定和上市都能提供科学的建议和帮助。
4.3.5 SEMs额外激励 我国SEMs普遍存在创新能力薄弱和研发资金不足的情况。建议在研发资助、协议帮助方面对SEMs进行一些倾斜。
4.3.6 市场独占 给予罕用药一定期限的市场独占是罕用药研发激励的核心措施,也是最强有力的措施。前文所述的措施都是在罕用药上市之前发挥作用的,而市场独占则是罕用药上市之后获得的一定时期的市场垄断,直接决定了罕用药上市之后的利润和预期回报,也是医药企业最重视的。市场独占首先需要确定的是独占期,要基于罕用药研发成本和预期收益设定合理的期限。其次,建议参考欧盟的经验,对独占期进行灵活处理,例如当该药品已经不符合罕用药的标准,或者销售收入超过某一标准后取消或缩短独占期。对于全新的罕用药和“老药新用”的罕用药在独占期的设定上也可以区别对待。最后,还要规定打破市场独占的情况,从临床优越性、是否征求了独占人的许可和市场供求角度来考虑。我国的新药监测期制度,规定获得生产许可之日起2年之内如果不生产,则取消监测期。为了和此制度接轨,也可以设立2年不生产则取消市场独占的规定。4.3.7 特殊审批 我国虽然已经建立了罕用药上市的特殊审批制度,但是具体程序还有待改善:一是设置多样化的审批程序,根据不同情况设立不同的特殊审批程序;二是明确特殊审批需要满足的技术标准和要点,特别是上市终点指标;三是明确特殊审批的药品上市后的风险效益评价工作。同美国和欧盟一样,特殊审批程序应该是我国药品注册改革的一项重要内容,不仅适用于罕用药。
4.4 加强与其他国家的合作
目前,在罕用药的资格认定和研发激励方面,美国与欧盟、日本已经建立了合作关系。我国虽然短期之内不太可能在资格申请、协议帮助、年度报告等方面和这些国家或地区马上建立合作和联系,但是可以做好国际合作的准备,以利于促进我国医药企业占据国际罕用药市场。
综上所述,我国应该尽快确定罕用药研发激励的相关立法、设立罕用药的资格认定、从多方面入手完善罕用药研发激励具体措施,同时加强与其他国家在罕用药资格认定和研发激励方面的合作。
[1]Gamm ie T,Lu CY,Babar ZU.Access to orphan drugs:a comprehensive review of legislations,regulationsand policies in 35 countries[J].PLoSOne,2015,10(10):e0140002.
[2]Braun MM,Farag-El-Massah S,Xu K,etal.Emergency of orphan drugs in the United States:a quantitative assessmentof the first25 years[J].NatRev Drug Discov,2010,9(7):519-522.
[3] Ogbah R,Associate L,Pharmalink G.Orphanmedicinal products:a European process overview[J].Regulatory Rapporteur,2015,12(2):5-11.
[4] FDA.Search orphan drug designations and approvals [EB/OL].(2016-06-25)[2016-07-15].http://www.accessdata.fda.gov/scripts/opdlisting/oopd/index.cfm.
[5] EMA.Orphan medicines figures:2000-2015[EB/OL].(2016-03-03)[2016-07-06].http://www.ema.europa. eu/docs/en_GB/document_library/Other/2015/04/ WC500185766.pdf.
[6] FDA.Orphan products grants program[EB/OL].(2016-01-13)[2016-07-16].http://www.fda.gov/ForIndustry/ DevelopingProductsforRareDiseasesConditions/Whom to-ContactaboutOrphanProductDevelopment/default.htm.
[7] Fellows GK,Hollis A.Funding innovation for treatment for rare diseases:adopting a cost-based yardstick approach [J].Orphanet JRare Dis,2013,8(1):180-185.
[8] Evaluate Pharma.Orphan drug report 2015[EB/OL].(2016-01-30)[2016-07-05].http://info.evaluategroup. com/rs/607-YGS-364/images/EPOD15.pdf.
[9] MurakamiM.Matched analysis on orphan drug designations and approvals:cross regional analysis in the United States,the European Union,and Japan[J].Drug Discov Today,2016,21(4):544-550.
[10] Galati F,BigliardiB.The unintended effectof the Orphan Drug Act on the adoption of open innovation[J].Sci Public Policy,2016,doi:10.1093/scipol/scw001.
Com parative Study and Enlightenment of the R&D Incentive Policies for Orphan Drugs in USA and the EU
YANG Li,TIAN Lijuan,LIN Lin(College of Business Adm inistration,Shenyang Pharmaceutical University,Shenyang 110016,China)
OBJECTIVE:To provide references and suggestions for building and improving the R&D incentive policies for orphan drugs in China.METHODS:The R&D incentive policies for orphan drugs in USA and the EU were compared in aspects of its legislative history,incentivemeasures and effects.And suggestions for improving related policies in China were put forward.RESULTS&CONCLUSIONS:The R&D incentive policies for orphan drugs in USA and the EU respectively started from Orphan Drug Act in USA(1983)and Orphan Drug Management Specification in the EU(1999),then formed relatively complete system by continuous improvement.The USA and the EU showed differences in its certification standard,procedure and specific incentives [R&D funding,tax deduction,fee reduction,additional incentives for micro and small and medium enterprises(SMEs),market exclusivity and special approval procedure],etc.In terms of fee reduction,for example,prescription application fee,production cost and drug confirmation fee were exempted in USA,while arrangement assist fee,initial and follow-up fee,checking fee before approval,initial listing type were reduced to a certain percentage in the EU.After developing incentive policies for orphan drugs,there is great increase in numbers of recognized qualifications and listing,SMEs have become new force in orphan drug R&D,R&D investment covers all types of diseases,orphan drug R&D are becoming themain direction of drug innovation and biotechnology development.China should determ ine the relevant legislation of R&D incentives for orphan drugs as soon as possible,set certification and improve specific measures of R&D incentives for orphan drugs from multiple aspects,while strengthen the cooperation w ith other countries in qualification and R&D incentives.
Orphan drug;Research and development incentive;USA;EU;Comparative;Research
R95
A
1001-0408(2017)16-2161-06
2016-10-12
2017-04-06)
(编辑:刘明伟)
国家社会科学基金项目(No.13CFX086);辽宁省教育厅科学研究一般项目(No.W 2014119);沈阳药科大学中青年教师事业发展支持计划
*副教授,博士。研究方向:药事法规与药物政策。E-mail:yanglishanxi@126.com
DOI10.6039/j.issn.1001-0408.2017.16.01
