SPE-GC-MS/MS法测定茶叶中49种农药残留
2017-07-01王吉祥牛之瑞冯雷白祥杜四淮谭建林张晓红
王吉祥,牛之瑞,冯雷,白祥,杜四淮,谭建林,张晓红
(云南省产品质量监督检验研究院,云南昆明650223)
SPE-GC-MS/MS法测定茶叶中49种农药残留
王吉祥,牛之瑞,冯雷,白祥,杜四淮,谭建林,张晓红
(云南省产品质量监督检验研究院,云南昆明650223)
建立固相萃取-气相色谱-三重四极杆串联质谱法测定茶叶中49种农药残留的分析方法。样品经乙腈均质提取,经ENVI-Carb+Florisil固相萃取小柱进行净化后,采用气相色谱-三重四极杆串联质谱法在多反应监测(MRM)模式下测定,外标法定量。49种农药成分在10μg/L~100μg/L范围内有良好的线性关系,相关系数在0.995 2~0.999 9范围内;49种农药成分在3个添加水平下(10、50、100 μg/L),加标回收率范围为71.43%~115.21%,RSD(n=6)为1.13%~11.05%;各农药成分的检出限均小于1.06 μg/kg。该方法样品处理简单快速,灵敏度和选择性高,适用于日常检测工作。
茶叶;气相色谱-三重四级杆串联质谱法;农药残留
云南是我国古老的茶区,茶产业是云南省的民生产业,同时茶叶是我国主要的经济作物,也是我国主要出口农产品之一。近年来,欧盟以及日本等发达国家对进口茶叶的农药残留检测项目不断增加,对限量标准的要求也日趋严格[1-3]。茶叶的安全卫生状况,特别是农药残留问题,始终是全世界关注的大问题,其分析技术的提高同样受到了高度重视。
近年来,对茶叶中农药残留的分析方法有不少报道,色谱-质谱联用法以其准确、灵敏等特点逐步成为茶叶中农药残留检测最常用的方法。在近两年发布的茶叶中农药残留检测方法的国家标准中,也主要采用了气相色谱-质谱法、液相色谱-串联质谱法。目前国标中尚无用气相色谱串联质谱法检测茶叶中多种农药残留的分析方法[4-6]。
茶叶中含有数百种化学成分,基质相对复杂,常用的色谱方法易受基质干扰,且易产生假阳性结果[7]。气相色谱-三重四级杆串联质谱的优势在于其具有极高的选择性、极强的抗干扰能力、高灵敏度和高通量离子传输效率及准确定量的特点,这使得该技术在复杂基质背景下仍能完成目标化合物的准确鉴定。采用多反应监测(multiple reaction monitoring,MRM)模式的串联质谱仪(tandem mass spectrometry,MS/MS)正在迅速成为复杂基质样品中多残留目标化合物筛查分析的理想技术,在食品安全分析中具有非常广泛的应用。本文建立了固相萃取-气相色谱-三重四极杆串联质谱法测定茶叶中49种农药残留。该方法简便、准确、快速、实用,各项技术指标满足检测要求,能够很好的应用于茶叶农药残留的测定。
1 材料与方法
1.1 材料、试剂与仪器
GCMS-TQ8030气相色谱-三重四级杆串联质谱仪:日本Shimadzu公司;GL-12B高速离心机:上海安亭科学仪器厂;N-EVAP氮吹仪:美国Organomation公司;CPA225D电子天平:德国Sartorius公司;十二孔固相萃取装置:美国Waters公司。
ENVI-Carb固相萃取小柱(3 mL)、Florisil固相萃取小柱(6 mL):美国Supelco公司;丙酮、乙腈、正己烷(均为色谱纯):美国Fisher Scientific公司。
普洱茶、绿茶、红茶、乌龙茶、白茶由云南省产品质量监督检验研究院提供。
49种农药标准品均购自农业部环境保护科研监测所,质量浓度为100 μg/mL。
标准溶液的配制:分别准确吸取1.0 mL上述标准品,用正己烷定容至10 mL,配成10.00 mg/L的标准储备液。根据实际需要,梯度稀释成系列浓度的标准工作溶液。
1.2 试验方法
1.2.1 样品制备
准确称取制好的茶叶粉末5.0 g,加入20.0 mL乙腈,超声30 min,5 000 r/min离心10 min,吸取2 mL上清液,低温氮吹至净干,用2.0 mL正己烷∶丙酮(2∶1,体积比)溶解,待净化。
将ENVI-Carb小柱用2.0 mL正己烷∶丙酮(2∶1,体积比)预淋洗,条件化,当溶剂液面到达柱吸附层表面时,立即倒入上述待净化溶液,用10 mL比色管接收洗脱液约8 mL,低温氮吹至约1 mL,用正己烷∶丙酮(9∶1,体积比)2 mL溶解,待下一步净化。
然后将Florisil小柱用2.0 mL正己烷∶丙酮(9∶1,体积比)预淋洗,条件化,当溶剂液面到达柱吸附层表面时,立即倒入上述待净化溶液,用10 mL比色管接收洗脱液约8 mL,低温氮吹至净干,用正己烷定容至0.5 mL,待测[8-9]。
1.2.2 色谱条件
色谱柱:Rtx-5MS(0.25 mm×0.25 μm×30 m)石英毛细管柱;载气:氦气,纯度≥99.999%;进样口温度:250℃;柱温:初始温度50℃,保持1 min,然后以25℃/min程序升温到125℃,再以10℃/min程序升温到300 ℃,保持 15 min;柱流速:1.69 mL/min;进样量:1 μL;进样方式:不分流进样,1 min后开阀。
1.2.3 质谱条件
电子轰击电离源(EI源)电压为70 eV;离子源温度200℃;接口温度250℃;溶剂延迟时间1.5 min;碰撞气:氩气(纯度≥99.999%);岛津GC Solution工作站。49种农药成分的保留时间及优化的MRM参数见表1。
2 结果与讨论
2.1 提取溶剂的选择[10-15]
茶叶中含有大量色素,特别是普洱茶、红茶中色素含量更高,且同时检测的农药品种数量较多,极性差异较大,因而对样品提取溶剂的选择要求较高。本文采用乙酸乙酯、乙腈、丙酮、二氯甲烷作为提取溶剂进行试验,结果发现,二氯甲烷溶出的色素和油脂较少,但不能充分提取所有农药;丙酮和乙酸乙酯能充分提取出大多数农药,同时也溶出大量的油脂和色素,增加了净化步骤和难度;乙睛作为极性较强的溶剂对大多数农药都能有效提取,且油脂和色素溶出率低,基质干扰小,回收率理想,乙腈与净化时的SPE柱相匹配,无需浓缩可直接过柱,操作简便。因此本试验采用乙睛作为提取溶剂。
2.2 固相萃取柱的选择
参照现行农药残留测定有关国家标准方法及国内外相关文献[10-15],本文比较了常用于农药残留的几种固相萃取柱:ENVI-carb+NH2柱、ENVI-carb+PSA柱和ENVI-carb+Florisil柱,结果ENVI-Carb+Florisil柱的净化效果最好,供试品溶液颜色最浅,总离子流中干扰峰较少,回收率最好,基质效应也较低,最终选用ENVI-Carb/Florisil固相萃取柱作为固相萃取净化方法。本方法采用ENVI-carb固相萃取柱作为固相萃取净化方法。见图1。
2.3 质谱条件的优化
本方法选择MRM下的2对离子对进行各农药的分析,其中1对离子对用于定量分析。为了获得最佳的质谱条件以保证对分析物定量和定性的准确性,对待测物的前级离子、产物离子、碰撞能量等一系列质谱参数进行了优化。首先采用全扫描(Full Scan)方式获取待测物的前级离子,之后采用产物离子扫描方式(Product Scan)通过优化碰撞能量获得产物离子,最后采用MRM模式对待测物进行定性和定量分析。经过方法优化可以得到较为理想的质谱条件和分离效果。每种农药的离子对和碰撞能,优化后见表1。图2为优化条件下10 μg/mL基质加标工作液的总离子流(TIC)色谱图。

图1 49种农药三种净化方法的回收率Fig.1 Recoveries of 49 pesticide residues obtained by three Purification methods

图2 49种农药的总离子流色谱图Fig.2 Chromatogram of MRM for 49 pesticide residues

表1 49种农药成分的保留时间及优化的MRM参数Table 1 Retention time and optimized MRM conditions of 49 pesticides
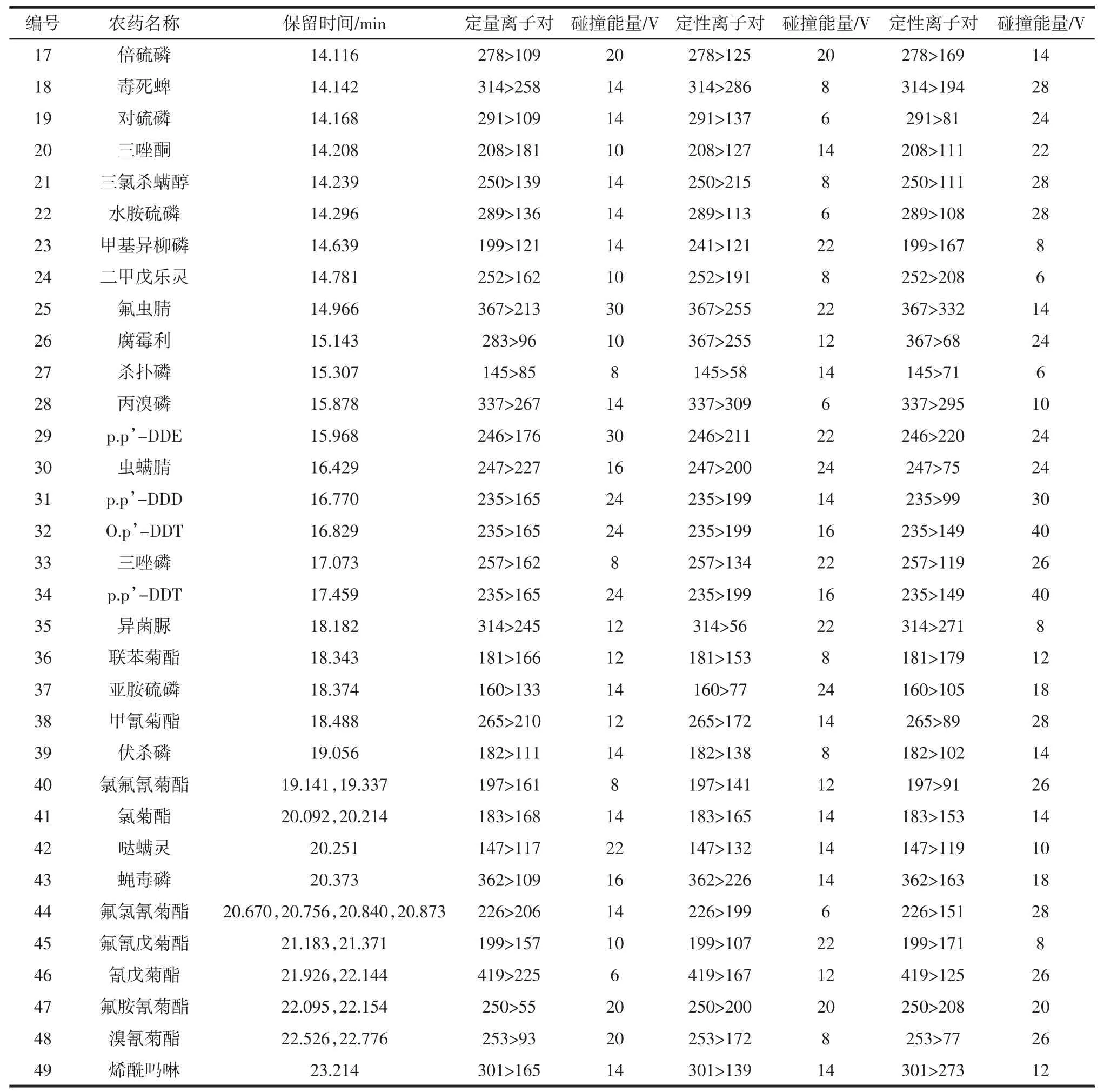
续表1 49种农药成分的保留时间及优化的MRM参数Continue table 1 Retention time and optimized MRM conditions of 49 pesticides
2.4 基质效应
茶叶成分复杂,对于农药残留的检测也有一定的影响,有些可能有基质增强作用,有些可能有基质减弱效应。对于气质农药残留分析,通常会出现基质增强效应,具体表现为使用农药标准品定量时会出现某种农药的回收率低于或高于可接受范围。为了避免药材本身带来的基质效应,采取基质混合标准品溶液的方法消除和减弱基质效应,同时利用选择离子定量检测也可以大大提高分析方法的选择性,排除一些杂质的干扰,最终获得较满意的试验结果。
2.5 线性关系与检出限
在质量浓度为10 μg/L~100 μg/L范围内,用基质配制成5个浓度的混合标准品系列溶液,在所确定的色谱条件下进行测定,分别对49种农药成分的峰面积对其浓度进行线性回归,建立标准曲线,以S/N≥3确定每种农药成分的检出限。结果表明:所测定的49种混合农药的质量浓度与对应的峰面积呈现良好的线性关系,相关系数(r2)均大于0.99,各农药成分的检出限均小于1.06 μg/kg,具体结果见表2。

表2 49种农药的线性方程、线性相关系数、检出限以及加标回收率和精密度Table 2 Linear equations,correlation coefficients(R2),limits of detection,the average recoveries and the relative standard deviations(RSD)for 49 pesticides
2.6 添加回收率和精密度
将农药混合标准贮备液添加到茶叶提取物中,分别制成质量浓度为 10、50、100 μg/kg的加标样品溶液,在同一分析条件下进行6次试验,计算各浓度下平均回收率及RSD。结果表明:49种农药成分在3个添加水平下(10、50、100 μg/L),加标回收率范围为71.43%~115.21% ,RSD(n=6)为 1.13%~11.05%,方法的回收率和相对标准偏差满足多农药残留检测中的准确度和精密度的要求,测定结果见表2。
2.7 方法适用性
采用本方法对100个茶叶样品(普洱茶30个、红茶20个、绿茶20个、乌龙茶15个、白茶15个)进行了分析测定,仅有联苯菊酯1种农药检出,含量在0.010 mg/kg~0.078 mg/kg之间(均为普洱茶),GB2763-2014《食品安全国家标准食品中农药最大残留限量》规定茶叶中联苯菊酯的最大残留限量分别为5 mg/kg,检出的农药残留水平均显著低于国家最大残留限量标准。
3 结论
试验通过优化检测条件,利用气相色谱三重四极杆质谱仪,建立了对茶叶中49种农药同时进行分析的方法。本方法条件下49种农药完全基线分离,每种化合物选择特征离子的两个二级离子,降低干扰,提高了灵敏度,特别对于复杂基质中假阳性结果进行排除。49种农药成分在10 μg/L~100 μg/L范围内有良好的线性关系,相关系数在0.995 2~0.999 9范围内;49种农药成分在 3 个添加水平下(10、50、100 μg/L),加标回收率范围为 71.43%~115.21%,RSD(n=6)为1.13%~11.05%;各农药成分的检出限均小于1.06 μg/kg。本分析方法获得了满意的分离效果和检测灵敏度,回收率、精密度,满足农药多残留分析的要求,检出限能满足欧盟及美国、日本等国的限量要求,该方法为茶叶中多农残测定提供了简便的前处理方法和检测手段。
[1] 岳永德,花日茂,张承祥.茶叶农药残留与控制[M].北京:中国农业出版社,2001:24-25
[2] 佚名.日本《食品中农业化学品肯定列表制度》对我国茶叶出口的影响及应对措施[J].农化市场十日讯,2006(17):11
[3] 中华人民共和国卫生部药典委员会.中华人民共和国药典[M].北京:化学工业出版社,2000:75
[4] 施家威,李继革,王玉飞,等.固相萃取-气相色谱/三重四级杆串联质谱分析蔬菜中42种农药残留[J].色谱,2010,28(12):1137-1143
[5] 沈伟健,余可垚,桂茜雯,等.分散固相萃取-气相色谱-串联质谱法测定蔬菜中107种农药的残留量[J].色谱,2009,27(4):391-400
[6] 孙磊丽,李晓玉,隋涛.气相色谱—串联质谱测定菠菜中有机氯残留量[J].广州化工,2010,38(11):155-157
[7] 易盛国,侯雪,韩梅,等.气相色谱-串联质谱法检测蔬菜农药残留基质效应与基质分类的研究[J].西南农业学报,2012,25(2):537-543
[8]中华人民共和国农业部.NY/T 761-2008蔬菜和水果中有机磷、有机氯、拟除虫菊酯和氨基甲酸酯类农药多残留检测方法[S].北京:中国农业出版社,2008
[9]中华人民共和国国家质量监督检验检疫总局,中国国家标准化管理委员会.GB/T 19648-2006水果和蔬菜中500种农药及相关化学品残留量的测定气相色谱-质谱法[S].北京:中国标准出版社,2006
[10]王连珠,周昱,陈泳,等.QuEChERS样品前处理-液相色谱-串联质谱法测定蔬菜中66种有机磷农药残留量方法评估[J].色谱,2012,30(2):146
[11]宋淑玲,李重九,马晓东,等.蔬菜中残留农药的石墨化碳黑净化和气相色谱-质谱检测方法[J].分析化学,2008,36(11):23
[12]LEHOTAY SJ,DE KOK A,HIEMSTRA M,et al.Validation of a fast and easy method for the determination of residues from 229 pesticides in fruits and vegetables using gas and liquid chromatography and mass spectrometric detection[J].JAOAC Int,2005,88(2):595
[13]ANASTASSIADES M,LEHOTAY SJ,STAJNBAHER D,et al.Fast and easy multiresidue method employing acetonitrile extraction/partitioning and dispersive solid-phase extraction for the determination of pesticide residues in produce[J].JAOAC Int,2003,86(2):412
[14]LEHOTAY S J,SON K A,KWON H.Comparison of QuEChERS sample preparation methods for the analysis of pesticide residues in fruits and vegetables[J].Journal of Chromatography A,2010,1217(16):2548-2560
[15]WILKOWSKA A,BIZIUK M.Determination of pesticide residues in food matrices using the QuEChERS Methodology[J].Food Chemistry,2011,125(3):803-812
Determination of 49 Kinds of Pesticide Residues in Tea by SPE and Gas Chromatography-Tandem Mass Spectrometry
WANG Ji-xiang,NIU Zhi-rui,FENG Lei,BAI Xiang,DU Si-huai,TAN Jian-lin,ZHANG Xiao-hong(Yunnan Product Quality Supervision and Testing Academy,Kunming 650223,Yunnan,China)
To develop an analysis method based on solid-phase extraction and gas chromatography-triple quadrupole tandem mass spectrometry(SPE-GC-MS/MS)or the determination of 49 kinds of pesticide residues in Tea.Samples were extracted with acetonitrile,and then purified through ENVI-Carb+Florisil solid phase extraction(SPE)cartridge,the prepared samples were then analyzed by GC-MS/MS in MRM mode,and the external standard method was applied to quantify the pesticides.All the 49 pesticides showed good linearity in the range of 10 μg/L to 100 μg/L (R2=0.995 2-0.999 9),the average recoveries of all the pesticides were in the range of 71.43%to 115.21%at three spiked levels of 10,50,100 μg/L,the RSD (n=6)were in the range of 1.13%to11.05%;and the limits of detection(LOD)were all below 1.06 μg/kg.The method is suitable for the multiresidues analysis of pesticides in Tea.
tea;gaschromatography-tandem massspectrometry(GC-MS/MS);pesticide residues
2016-10-09
王吉祥(1983—),男(汉),工程师,硕士,研究方向:农药残留检测。
10.3969/j.issn.1005-6521.2017.13.037
