CD133 epitope vaccine with gp96 as adjuvant elicits an antitumor T cell response against leukemia
2017-07-01ShuoWangHongxiaFanYangLiHuaguoZhengXinLiChangfeiLiLizhaoChenYingJuandSongdongMeng
Shuo Wang, Hongxia Fan, Yang Li, Huaguo Zheng, Xin Li, Changfei Li, Lizhao Chen, Ying Ju, and Songdong Meng,4
CD133 epitope vaccine with gp96 as adjuvant elicits an antitumor T cell response against leukemia
Shuo Wang1*, Hongxia Fan2*, Yang Li3, Huaguo Zheng3, Xin Li3, Changfei Li3, Lizhao Chen3, Ying Ju3, and Songdong Meng3,4
1 College of Life Sciences, Hebei University, Baoding 071002, Hebei, China 2 School of Basic Medical Sciences, Tianjin Medical University, Tianjin 300070, China 3 CAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China 4 College of Life Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
Cancer stem cells are currently under intensive investigation due to their capabilities for tumor initiation, self-renewal, and resistance to chemotherapy. CD133 is implicated in stemness and the malignancy of tumor cells. Here, we explored heat shock protein gp96 adjuvanted CD133 epitope vaccine against leukemia. We screened and identified three H2-Kd-restricted cytotoxic T lymphocyte (CTL) epitopes derived from CD133, CD133419-428, CD133702-710and CD133760-769. The immunogenicity and antitumor activity of the epitope vaccine using heat shock protein gp96 as adjuvant were further determined in CD133+leukemia xenograft mice. Finally, we demonstrate that adoptive transfer of epitope-specific CTLs led to suppression of leukemia growth. Our data therefore provide the basis for designing a CD133 epitope vaccine to activate specific CTLs against CD133+leukemia and other cancers.
CD133, epitope, gp96, leukemia, CTL
Leukemia is the most common blood cell cancer in the world. According to2016, there are 60 140 new cases of leukemia in the US that will lead to 24 400 deaths. Adults older than 50 years of age have susceptibility incidence rates for developing leukemia of 2.0% (men) and 1.2% (women). Leukemia is also the second leading cause of death from cancer among people younger than 20 years[1]. Several lines of evidence indicate that leukemia is initiated and sustained by a small population of cancer stem cells (CSCs)[2-4].
CD133, also known as AC133 or prominin-1, is a cell surface glycosylated protein that was first identified in human hematopoietic stem cells. It has five-transmembrane domains with a molecular weight of 120 kDa[5]. CD133 has been defined as a CSC surface antigen marker in various malignancies, including breast cancer, brain tumors, lung cancer, glioblastoma, prostate cancer, and colon cancer[6-11]. Although its physiological functions are not entirely known, CD133 may be involved in the regulation of cell proliferation through the WNT pathway, primitive cell differentiation, and epidermal- mesenchymal interactions[12-13]. Because CSCs are undifferentiated cells that can self-renew and overexpress stemness markers (, CD133), drug transporters, and metastasis-related proteins, it has been suggested that these malignant cells play a key role in tumor initiation and progression, as well as tumor resistance to conventional cytotoxic/anti- proliferative therapies, including chemotherapy and radiation-therapy[14].
The expression of CD133 is also detectable in patients with acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL). Further, it seems to be associated with shorter overall survival and poor prognosis, as well as higher resistance to standard chemotherapy and relapse[15]. CD133 expression is induced by the AF4 transcription factor and plays an essential role in leukemia cell survival and growth[16]. Moreover, imatinib therapy, an important first-line treatment option against chronic myeloid leukemia (CML) that targets the oncogene product BCR-ABL, cannot effectively eliminate CML stem cells[17-18]. Given that its pathways in leukemia and many other cancers possess broad functions in cancer initiation, metastasis, and drug resistance, CD133 is an ideal target for therapeutic development against cancer.
T cells, especially cytotoxic T cells (CTLs), possess essential features of successful immune-based strategies toward cancer. We previously demonstrated that similar to native heat shock protein gp96,-expressed recombinant gp96 as adjuvant induces a potent CTL immune response towards antigens that were complexed with it[19]. In this study, we screened potential T cell epitopes from CD133. Their immunological function and antitumor activity were further assessed using recombinant gp96 as adjuvant. Our study may provide the basis for designing an epitope-based vaccine against leukemia and CSCs.
1 Materials and methods
1.1 Ethics statement
Animal studies were performed according to the guidelines set forth by the Institute of Microbiology, Chinese Academy of Sciences of Research Ethics Committee. All animal experiments were performed in strict accordance with institutional guidelines on the handling of laboratory animals. Mice were euthanized when the maximum tumor size (diameter: 2.0 cm) had been reached.
1.2 Peptide synthesis
H2-Kd-restricted epitopes from mouse CD133 (prominin-1 isoform s8 precursor, Genomic sequence: NP_001157057.1) were predicted with SYFPEITHI (http://www.syfpeithi.de/), Bimas (http://www-bimas.cit.nih.gov/), and Immune epitope database and analysis resource (IEDB) (http://www.iedb.org/). Predicted CD133 epitopes CD133272–281(QNMSSSLKSL), CD133419–428(RYLNQELPKL), CD133452–461(FFFLGLLCGV), CD133601–609(NLNVNIDSI), CD133702–710(TLRQSVWTL), and CD133760–769(HYLHWVFYAI), as well as the negative control HBc18–27(FLPSDFFPSV) and positive control HBc87–95(SYVNTNMGL), peptides were synthesized by GL Biochem (Shanghai, China). Their purity (>95%) was confirmed by high-performance liquid chromatography (HPLC) and mass spectrometry.
1.3 Mice and cell culture
Female BALB/c (H2-Kd) and DBA/2 mice (H2-Kd) were purchased from Beijing Weitonglihua Laboratory Animal Technology Co., Ltd. (Beijing, China). All mice were 6−8 weeks of age at the start of all experiments and housed under pathogen-free conditions according to institutional guidelines.
Murine L1210lymphocytic leukemia cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (GIBCO) supplemented with 10% (/) heat-inactivated fetal calf serum, 25 μg/mL streptomycin, and 100 IU/mL penicillin and maintained in a 37 °C incubator with 5.0% CO2.
1.4 Immunization of mice
The-to-Expression System was used to express the recombinant heat shock protein mouse gp96 as described previously[19]. The endotoxin was tested using the quantitative chromogenic Limulus amebocyte lysate assay (QCL-1000; BioWhittaker, Walkersville, MD, USA), and its concentration was as low as 0.01 EU/μg recombinant gp96 protein. The gp96-CD133 peptide complexes were prepared by incubation of 20 μg gp96 with 50 μg peptide in binding buffer (20 mmol/L HEPES (pH 7.2), 20 mmol/L NaCl, and 2 mmol/L MgCl2) for 10 min at 50 °C. Then, the preparations were placed at room temperature for 30 min for the binding of gp96 to the peptides.
DBA/2 mice were subcutaneously immunized with gp96-CD133 peptide complexes, a mixture of gp96-CD133 peptides complexes, or gp96-control peptide complexes at weeks 1, 2, and 4 (respectively) or as described. In tumor prophylactic and therapeutic experiments, mice were subcutaneously challenged (in the right flank) with 5×104L1210 tumor cells. Tumor size was determined by measuring the smallest diameter (a) and the largest diameter (b) using calipers. Tumor volume () was calculated using the formula:=2/2.
1.5 Adoptive T cell transfer
Splenocytes were isolated from the spleens of immunized or unimmunized (control peptide) DBA/2 mice as described[20]. CD3+T cells were enriched (>90% purity) by magnetic bead separation (Miltenyi, Biotec, Germany) from cultured splenocytes. Purified T cells (2×107cells/mice) were intravenously injected into γ-irradiated (500 rads) recipient mice.
1.6 IFN-gELISPOT and enzyme-linked immunosorbent assay (ELISA) analysis
The IFN-γ ELISPOT assays were performed according to the manufacturer’s protocol. Briefly, 96-well PVDF plates (BD-Pharmigen, San Diego, CA) were pre-coated overnight at 4 °C with the coating antibody (Ab) and blocked for 1 h at 37 °C. Isolated murine splenocytes were placed in each well (1×106cells/well) in triplicate, and peptides (10 μg/mL) were added to the well and incubated at 37 °C for 48 h. The spots were counted and analyzed with an ELISPOT Reader (Biosys, Germany).
The levels of IFN-γ, IL-2, IL-4, TNF-α and IL-6 in culture supernatants were determined by ELISAs following the manufacturer’s protocol (eBioscience, San Jose, CA).
1.7 Intracellular cytokine staining
Splenocytes from immunized mice were stimulated with the peptides for 72 h, stained with anti-CD8 and IFN-γ, and analyzed by flow cytometry.
1.8 T cell cytotoxicity assay
L1210 cells were labeled with 5-(6)-carboxy fluorescein diacetate succinimidyl ester (CFSE) (Sigma-Aldrich) as target cells and seeded into a 96-well plate. Murine splenocytes stimulated with CD133 peptides or control peptide were served as effector cells and added at different effector-to-target ratios of 20:1, 10:1, 5:1 and 2.5:1. Plates were incubated for 4 h at 37 °C, and CFSE-positive cells were stained with propidium iodide (PI) using a Vybrant Apoptosis Assay Kit (Invitrogen, USA) and analyzed by flow cytometry. Cytotoxicity per well was calculated as the percentage of dead target cells. Each assay was performed in triplicate.
1.9 Statistical methods
2 Results
2.1 Identification of H2-Kd-restricted epitopes from murine CD133
The MHC class I epitope prediction algorithms SYFPEITHI, Bimas, and IEDB were used to predict potential CD8+T cell epitopes within the full-length mouse CD133 protein (aa 1–842) that bind H2-Kd. Six peptides had high ranking:CD133272–281(QNMSSSLKSL), CD133419–428(RYLNQELPKL), CD133452–461(FFFLGLLCGV), CD133601–609(NLNVNIDSI), CD133702–710(TLRQSVWTL), and CD133760–769(HYLHWVFYAI) (Fig. 1A). To confirm that these 9- to 10-mer peptides are Kd-restricted epitopes, BALB/c mice were subcutaneously immunized with the six synthesized peptides three times at weeks 1, 2 and 4, using recombinant heat shock protein gp96 as an adjuvant[21]. Peptide-specific CTLs from splenocytes were detected by IFN-γ ELISPOT assays 1 week after the last immunization. As shown in Figure 1B, CD133419–428, CD133702–710, and CD133760–769significantly induced CTL responses compared to the negative peptides. No peptide-specific CD8+T cell response was detected from CD133272–281, CD133452–461or CD133601–609peptide-immunized mice. This indicates that CD133419–428, CD133702–710and CD133760–769are Kd-restricted CTL epitopes.
2.2 CD133 epitopes generate specific CTL responses in Kd-restricted mice
Next, the immunogenicity of the CD133419–428, CD133702–710and CD133760–769epitopes was determined. BALB/c mice were subcutaneously vaccinated with CD133419–428, CD133702–710or CD133760–769peptide at weeks 1, 2 and 4, with gp96 as adjuvant. The three epitopes vaccine formulations effectively elicited T cell responses to CD133419–428, CD133702–710and CD133760–769peptides as judged by ELISPOT assays, while only very low peptide-specific T-cell immunity could be detected in control mice (Fig. 2A). As shown in Figure 2B, a dramatically increased number of IFN-γ producing CD8+T cells in the spleen were observed between mice treated with CD133 epitopes and control peptides (CD133 epitopescontrol: CD8+T cells, 4.34% ± 0.06%1.09% ± 0.29%,<0.01). In addition, splenocytes from mice vaccinated with CD133 epitopes had significantly higher specific cytolytic activity than mice vaccinated with control peptides in killing assays using L1210 lymphocytic leukemia cells as target cells (Fig. 2C). Furthermore, the supernatants of splenocytes stimulated with CD133 or control peptides were measured for the production of the cytokines IFN-γ, IL-2, TNF-α, IL-6 and IL-4 by ELISAs. As shown in Figure 2D, the levels of IFN-γ, IL-2, TNF-α, IL-6 and IL-4 from CD133 epitope-immunized mice were significantly higher than those from control mice (<0.01 or 0.05). No significant humoral and T cell responses against the adjuvant gp96 were observed in immunized mice (data not shown), indicating low immunogenicity of gp96 itself.

Fig. 1 Detection of CD133-derived peptide-specific CD8+ T cell responses in BALB/c mice. (A) Schematic representation of the amino acid positions of predicted H2-Kd-restricted epitopes from mouse CD133 protein. (B) Peptide-specific CTLs were detected by IFN-γ ELISPOT assays. CD133272–281, CD133419–428, CD133452–461, CD133601–609, CD133702–710 or CD133760–769 peptides were complexed with recombinant gp96 protein in vitro, respectively. BALB/c mice were immunized with gp96-CD133 peptide complexes three times at weeks 1, 2 and 4, or with gp96-HBc18–27 or gp96-HBc87–95 complexes as the negative and positive controls, respectively (n=5 mice/group). * P<0.05; ** P<0.01 compared to the control. Similar results were observed in three independent experiments.

Fig. 2 CD133 epitopes induce specific CTL responses in BALB/c mice. BALB/c mice were immunized with CD133419–428, CD133702–710 and CD133760–769 peptides (50 μg each/mice) complexed with gp96 (20 μg per mice) three times at weeks 1, 2 and 4, or with gp96-HBc18–27 complexes (150 μg per mice) as a control (n=5 mice/group). Mice were sacrificed at week 5, and splenocytes were isolated for analysis. (A) Peptide-specific CTLs were detected by IFN-γ ELISPOT assays. Splenocytes (1×106 cells/well) were stimulated with 10 μg/mL of CD133419–428, CD133702–710, CD133760–769 or HBc18–27 peptide. (B) FACS analysis to quantify IFN-γ+CD8+ T cell populations in mouse spleens. (C) Cytotoxicity assay. Splenocytes from CD133 epitopes or control peptide immunized mice were incubated with a mixture of CD133419–428, CD133702–710 and CD133760–769 peptides, or control peptide for 72 h and analyzed for cytotoxic activity using CFSE-labeled L1210 cells as target cells. PI staining was then used to measure cell lysis by FACS analysis. The killing activity is indicated as the mean percentage of specific lysis (± s.d.) at different effector-to-target (E:T) ratios. (D) Secretion of IFN-γ, IL-2, IL-4, TNF-α and IL-6 by splenocytes stimulated with peptides for 48 h was determined by ELISA assays. *P<0.05; **P<0.01 compared to the control. Similar results were observed in three independent experiments.
2.3 CD133 epitopes elicit antitumor T cell responses against murine leukemia
Next, we examined whether CD133 epitopes are able to induce antitumor T cell responses using a murine leukemia model. Expression of cell membrane CD133 in lymphocytic leukemia L1210 cells was confirmed by FACS analysis (Fig. 3A). Heterogeneous expression of CD133 on L1210 cells was observed as the histogram of CD133 showed two peaks, which indicates that there may exist CD133low- and CD133high-expressing L1210 cell subsets. The prophylactic efficiency of CD133 epitopes was first examined. DBA/2 mice were subcutaneously immunized with the combination of gp96-CD133419–428, -CD133702–710and -CD133760–769complexes or control peptide three times and challenged with L1210 leukemia cells (5×104cells/mouse) 1 week after the third immunization. Compared to control peptide, immunization with CD133 epitopes significantly inhibited tumor growth, decreasing tumor volume and weight both by approximately 43% (both<0.05) (Fig. 3B and 3C). As seen in Figure 3D, ELISPOT assays revealed that immunization with CD133 epitopes resulted in about 4.6-, 0.6- and 1.2-fold increases in tumor-specific T cells compared to control peptide immunization, and only weak CTL response was observed in control group.In addition, splenocytes from CD133 epitopes-immunized mice exhibited significantly higher cytotoxicity activity towards L1210 cells than those from control peptide-immunized mice (Fig. 3E).
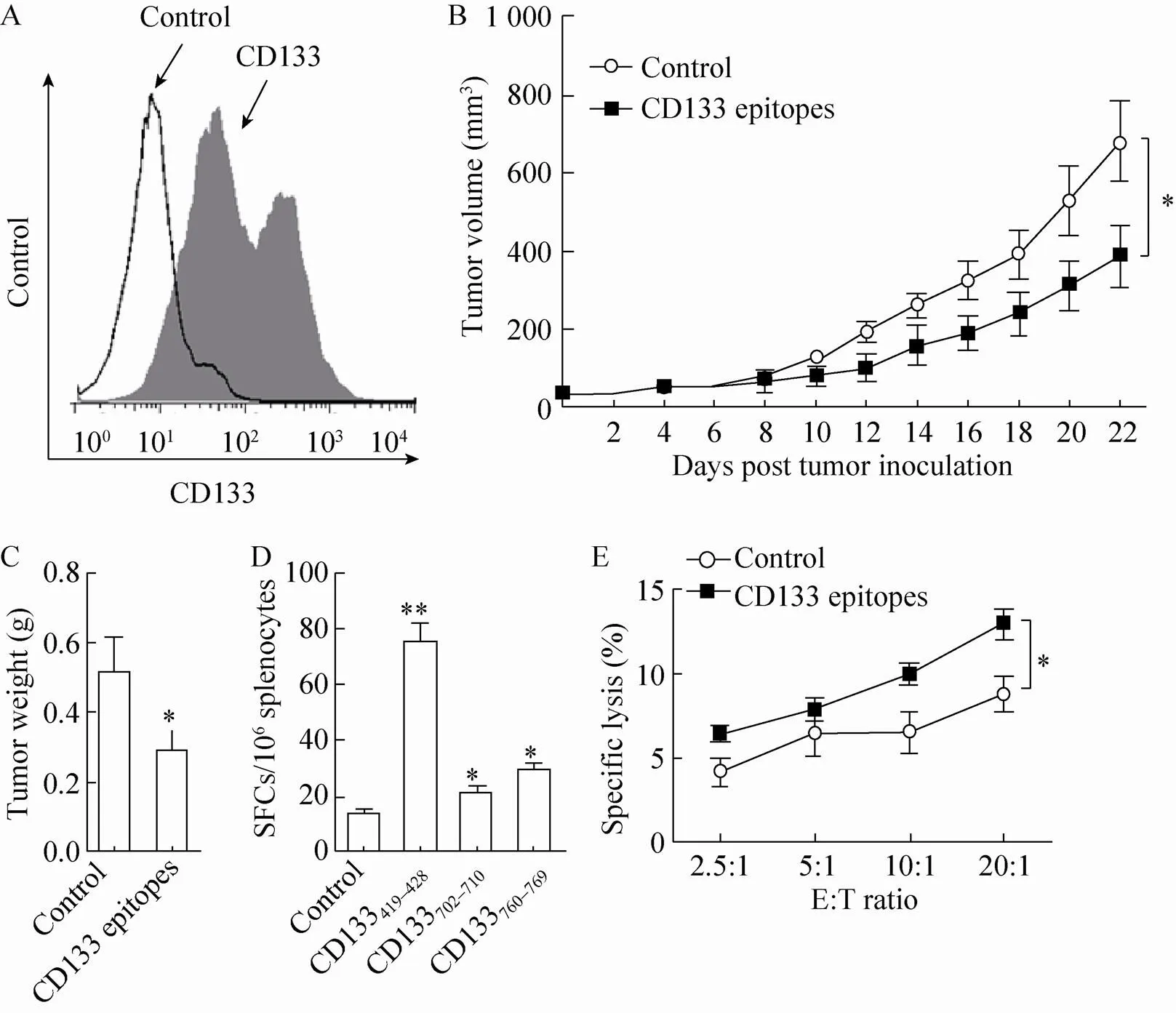
Fig. 3 Immunization with the CD133 epitope vaccine induced a prophylactic antitumor T cell response. (A) Flow cytometric analysis of cell surface CD133 levels in L1210 cells. (B-E) DBA/2 mice were immunized three times with gp96-CD133419–428, gp96-CD133702–710 or gp96-CD133760–769 complexes (50 μg per mice), or with 150 μg gp96-HBc18-27 complex as a control (n=5 mice/group). One week after the third immunization, the mice were subcutaneously challenged with 5×104 L1210 cells/mice. Tumor burden was measured at 2-day intervals (B). Mice were sacrificed at week 3 post tumor challenge, and tumor weight was measured (C). Splenocytes from CD133 epitopes or control peptide immunized mice were stimulated with CD133419–428, CD133702–710 or CD133760–769 peptides for 48 h, respectively, or control peptide gp96-HBc18–27 for background evaluation, and assayed by IFN-γ ELISPOT assays (D). Splenocytes from CD133 epitopes or control peptide immunized mice were incubated with a mixture of CD133419–428, CD133702–710 and CD133760–769 peptides or control peptide for 72 h and analyzed for cytotoxic activity using CFSE-labeled L1210 cells as target cells (E). * P<0.05, ** P<0.01 compared to the control. Similar results were observed in two independent experiments.
To determine the therapeutic efficiency of the CD133 epitope vaccine, DBA/2 mice were injected with L1210 cells (5×104cells/mice), and after 1 week (tumor size around 100 mm3), mice were immunized with gp96-adjuvanted CD133 epitope vaccine three times. Compared to controls, immunization with CD133 epitope vaccine significantly inhibited tumor growth (Fig. 4A) and decreased tumor weight (Fig. 4B) by approximately 31 and 30%, respectively (both<0.05). Strong T cell-mediated cytotoxic activity against L1210 cells was observed in mice immunized with CD133 epitope vaccine (<0.01) (Fig. 4C). Together, these results indicate that gp96-based CD133 epitope vaccines can efficiently stimulate specific T cell responses and inhibit tumor growth.
2.4 Suppression of tumor growth by adoptive transfer of T cells from CD133 epitope-immunized mice
Finally, DBA/2 mice were immunized with gp96-adjuvanted CD133 epitope vaccine three times, and CD3+T cells were isolated from the spleen 1 week after the third immunization. As seen in Figures 5A and 5B, adoptive transfer of CD3+T cells from CD133 epitope vaccine-immunized but not control peptide-immunized mice resulted in significant inhibition of tumor growth (Fig. 5A) and decreased tumor weight (Fig. 5B) in irradiated recipient mice challenged with L1210 cells. This indicates that specific T cells play a key role in CD133 vaccine-induced tumor inhibition.
3 Discussion
CD133 expression is frequently observed in a subset of leukemia cells and is associated with tumor malignancy in patients. In this study, we screened and identified three CD133-specific CTL epitopes: CD133419–428, CD133702–710and CD133760–769. Moreover, we demonstrated that the CD133 epitope-based vaccine using heat shock protein gp96 as adjuvant exhibits antitumor activity in leukemia xenograft mice. Adoptive transfer experiments revealed that CD133-specific CTLs could effectively suppress tumor growth. Our work may therefore aid in the design of a CD133 epitope vaccine to activate specific CTLs against leukemia and other cancers, as well as provide the basis for using gp96 as a T cell adjuvant in boosting T cell responses against CD133-positive tumors and CSCs.

Fig. 4 Immunization with the CD133 epitope vaccine induced a therapeutic antitumor T cell response. DBA/2 mice were subcutaneously challenged with 5×104 L1210 cells/mice. Mice were immunized three times at 3-day intervals with CD133 epitope vaccine 1 week after tumor inoculation when tumors reached a volume of ~100 mm3 (n=5 mice/group). Tumor size was measured at 2-day intervals (A). Mice were sacrificed at week 3 post tumor challenge, and tumor weight was measured (B). Splenocytes from CD133 epitopes or control peptide immunized mice were isolated and stimulated with a mixture of CD133419–428, CD133702–710 and CD133760–769 peptides or HBc18-27 for 72 h and analyzed for cytotoxic activity by FACS using CFSE-labeled L1210 cells as target cells (C). **P<0.01 compared to the control. Similar results were observed in two independent experiments.

Fig. 5 Growth of L1210 tumors after adoptive transfer of CD3+ T cells from CD133 vaccine-immunized mice into irradiated recipient DBA/2 mice. Purified T cells from CD133 epitope vaccine- or control peptide-immunized DBA/2 mice were intravenously injected into γ-irradiated recipient mice, followed by subcutaneous challenge with 5×104 L1210 cells/mouse (n=5 mice/group). Tumor size was measured at 2-day intervals (A), and mice were sacrificed 3 weeks post tumor inoculation for measurement of tumor weight (B). **P<0.01 compared to the control. Similar results were observed in two independent experiments.
In current study, three H2-Kd-restricted CTL epitopes from CD133 were defined. The CD133 protein contains five-transmembrane domains. Interestingly, prediction of transmembrane domains using TMpred (http://www.ch.embnet.org/software/ TMPRED_form.html) shows that these three defined epitopes are all located in CD133 extracellular domains. This may facilitate epitope presentation and recognition by CD8+T cells.
CSCs were first identified in human acute myeloid leukemia (AML) cells by transplanting AML-initiating cells into severe combined immune-deficient (SCID) mice, and rare tumor populations within the tumor could form new abnormal tissues with indefinite potential for self-renewal[2, 22-23]. Now, compelling evidence shows that CSCs are responsible for tumorigenicity, tumor cell growth and anti-apoptosis, invasion, recurrence, and increased radioresistance and chemotherapy resistance in multiple cancers, including colon cancer[24-25], glioblastoma[9], pancreatic cancer[26], prostate cancer[10], lung cancer[8], breast cancer[27-28]and ovarian cancer[29-30]. In this regard, uncovering specific targets and eradicating these tumor-forming cells may provide a more efficient way to develop cancer treatments. Among the CSC markers, CD133 is expressed in a subpopulation of different tumors that has progenitor properties of tumor formation and aggressive invasion. Therefore, targeting CD133 with a monospecific anti-CD133 antibody conjugated to a toxin, anti-CD133/CD3 bispecific antibody, chimeric antigen receptor (CAR) T therapy, or siRNA delivery using nanocarriers represent attractive strategies for cancer therapy in both cell and animal models[13-14,31-35]. In this study, we identified three new CD133 epitopes that elicit strong CTL responses in mice. Furthermore, immunization with the CD133 epitope-based vaccine inhibited CD133+leukemia tumor growth in mouse xenograft models, suggesting that CD133 is a valid target for CTLs.
CD8+T cells are the main effector cells responsible for tumor cytolysis by both direct contact-mediated cytotoxicity and secretion of antitumor effector cytokines. The development of tumor-specific antigen/adjuvant or dendritic cell/T cell-based immunotherapies to stimulate effective innate and T cell immunity against cancer represents a major effort in cancer immunotherapy. In this study, we used recombinant gp96 as a T cell adjuvant for the CD133 epitope vaccine. As a chaperone in the endoplasmic reticulum, gp96 constitutes a relay line for loading degraded cellular peptides onto calreticulin and MHC molecules in a concerted and regulated manner for T cell activation[36-37]. In addition, gp96 itself interacts with and activates Toll-like receptors (TLRs), and elicits the innate immune response[38-39]. Our recent studies demonstratethator-expressed recombinant gp96 has capacity for peptide association, presentation, TLR activation, and induction of strong cellular immunoresponses, making recombinant gp96 a promising candidate for designing and engineering effective vaccines aimed at eliciting T-cell responses for prophylactic and therapeutic applications[19,21]. Our current findings indicate that recombinant gp96 could efficiently induce CTL responses against CD133+tumors, further validating the immunological potency of recombinant gp96 as a T cell adjuvant.
Our current study only provides a proof-of-principle for using a gp96-based CD133 epitope vaccine against CD133+leukemia. Further experiments are needed to validate its efficiency against other tumors where CD133+CSCs frequently constitute a small minority of the overall bulk tumor population. In addition, it would be worthwhile to investigate whether the CD133 epitope vaccine will stimulate T cell immune responses against non-neoplastic normal body cells that express comparable levels of CD133, including normal hematopoietic, neuronal, and endothelial progenitors[13].
In current study, we only show that the complexes of gp96-three CD133 peptides combination have promising antitumor activityand, as polyvalent vaccines are likely to initiate broader immune responses and less susceptible to tumor immune escape compared to monovalent vaccines[40-42]. The antitumor immune activity of each single peptide needs to be determined and compared in our future studies.
In summary, this study indicates that the defined Kd-restricted epitopes could elicit potent CTL responses. We further demonstrated that a CD133 epitope vaccine using recombinant gp96 as adjuvant has potent antitumor immune activity against CD133+leukemia in mice. Given the key role of CD133+CSCs in cancer development and metastasis, our work provides valuable insight into the functional implications of CD133 epitope-specific T cell responses in leukemia and developed an immunotherapeutic vaccine against cancer.
[1] Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin, 2016, 66(1): 7–30.
[2] Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature, 1994, 367(6464): 645–648.
[3] Guo W, Lasky JL, Chang CJ, et al. Multi-genetic events collaboratively contribute to-null leukaemia stem-cell formation. Nature, 2008, 453(7194): 529–533.
[4] Zhou H, Xu RZ. Leukemia stem cells: the root of chronic myeloid leukemia. Protein Cell, 2015, 6(6): 403–412.
[5] Yin AH, Miraglia S, Zanjani ED, et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood, 1997, 90(12): 5002–5012.
[6] Wright MH, Calcagno AM, Salcido CD, et al.breast tumors contain distinct CD44+/CD24-and CD133+cells with cancer stem cell characteristics. Breast Cancer Res, 2008, 10(1): R10.
[7] Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res, 2003, 63(18): 5821–5828.
[8] Eramo A, Lotti F, Sette G, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ, 2008, 15(3): 504–514.
[9] Bao SD, Wu QL, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature, 2006, 444(7120): 756–760.
[10] Collins AT, Berry PA, Hyde C, et al. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res, 2005, 65(23): 10946–10951.
[11] Arab-Bafrani Z, Shahbazi-Gahrouei D, Abbasian M, et al. Culturing in serum-free culture medium on collagen type-I-coated plate increases expression of CD133 and retains original phenotype of HT-29 cancer stem cell. Adv Biomed Res, 2016, 5(1): 59.
[12] Barcelos LS, Duplaa C, Kränkel N, et al. Human CD133+progenitor cells promote the healing of diabetic ischemic ulcers by paracrine stimulation of angiogenesis and activation of Wnt signaling. Circ Res, 2009, 104(9): 1095–1102.
[13] Schmohl JU, Vallera DA. CD133, selectively targeting the root of cancer. Toxins, 2016, 8(6): 165.
[14] Rassouli FB, Matin MM, Saeinasab M. Cancer stem cells in human digestive tract malignancies. Tumor Biol, 2016, 37(1): 7–21.
[15] Tolba FM, Foda ME, kamal HM, et al. Expression of CD133 in acute leukemia. Med Oncol, 2013, 30(2): 527.
[16] Mak AB, Nixon AM, Moffat J. The mixed lineage leukemia (MLL) fusion–associated genepromotes CD133 Transcription. Cancer Res, 2012, 72(8): 1929–1934.
[17] Branford S, Rudzki Z, Walsh S, et al. High frequency of point mutations clustered within the adenosine triphosphate–binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood, 2002, 99(9): 3472–3475.
[18] Corbin AS, Agarwal A, Loriaux M, et al. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest, 2011, 121(1): 396–409.
[19] Liu WW, Chen M, Li XH, et al. Interaction of toll-like receptors with the molecular chaperone Gp96 is essential for its activation of cytotoxic T lymphocyte response. PLoS ONE, 2016, 11(5): e0155202.
[20] Zhao B, Wang YZ, Wu B, et al. Placenta-derived gp96 as a multivalent prophylactic cancer vaccine. Sci Rep, 2013, 3: 1947.
[21] Li Y, Song HL, Li J, et al.expressed heat shock protein gp96 exerts potent T cell activation activity as an adjuvant. J Biotechnol, 2011, 151(4): 343–349.
[22] Fialkow PJ, Denman AM, Jacobson RJ, et al. Chronic myelocytic leukemia: origin of some lymphocytes from leukemic stem cells. J Clin Invest, 1978, 62(4): 815–823.
[23] Reya T, Morrison SJ, Clarke MF, et al. Review article stem cells, cancer, and cancer stem cells. Nature, 2001, 414(6859): 105–111.
[24] Todaro M, Alea MP, Di Stefano AB, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell, 2007, 1(4): 389–402.
[25] Vermeulen L, Todaro M, de Sousa Mello F, et al. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc Natl Acad Sci USA, 2008, 105(36): 13427–13432.
[26] Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell, 2007, 1(3): 313–323.
[27] Karnoub AE, Dash AB, Vo AP, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature, 2007, 449(7162): 557–563.
[28] Charafe-Jauffret E, Ginestier C, Iovino F, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res, 2009, 69(4): 1302–1313.
[29] Baba T, Convery PA, Matsumura N, et al. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene, 2009, 28(2): 209–218.
[30] Alvero AB, Chen R, Fu HH, et al. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle, 2009, 8(1): 158–166.
[31] Smith LM, Nesterova A, Ryan MC, et al. CD133/prominin-1 is a potential therapeutic target for antibody-drug conjugates in hepatocellular and gastric cancers. Br J Cancer, 2008, 99(1): 100–109.
[32] Canter RJ, Grossenbacher SK, Ames E, et al. Immune targeting of cancer stem cells in gastrointestinal oncology. J Gastrointest Oncol, 2016, 7(S1): S1-S10.
[33] Pan Q, Li Q, Liu S, et al. Concise review: targeting cancer stem cells using immunologic approaches. Stem Cells, 2015, 33(7): 2085–2092.
[34] Naujokat C. Monoclonal antibodies against human cancer stem cells. Immunotherapy, 2014, 6(3): 290–308.
[35] Zhu XK, Prasad S, Gaedicke S, et al. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget, 2015, 6(1): 171–184.
[36] Kropp LE, Garg M, Binder RJ. Ovalbumin-derived precursor peptides are transferred sequentially from gp96 and calreticulin to MHC class I in the endoplasmic reticulum. J Immunol, 2010, 184(10): 5619–5627.
[37] Messmer MN, Pasmowitz J, Kropp LE, et al. Identification of the cellular sentinels for native immunogenic heat shock proteins. J Immunol, 2013, 191(8): 4456–4465.
[38] Wu S, Hong F, Gewirth D, et al. The molecular chaperone gp96/GRP94 interacts with Toll-like receptors and integrins via its C-terminal hydrophobic domain. J Biol Chem, 2012, 287(9): 6735–6742.
[39] Yang Y, Liu B, Dai J, et al. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity, 2007, 26(2): 215–226.
[40] Tam JP. Synthetic peptide vaccine design: synthesis and properties of a high-density multiple antigenic peptide system. Proc Natl Acad Sci USA, 1988, 85(15): 5409–5413.
[41] Keenan BP, Jaffee EM. Whole cell vaccines—past progress and future strategies. Semin Oncol, 2012, 39(3): 276–286.
[42] Wagner SC, Riordan NH, Ichim TE, et al. Safety of targeting tumor endothelial cell antigens. J Transl Med, 2016, 14(1): 90.
(本文责编 陈宏宇)
CD133表位联合热休克蛋白佐剂疫苗引发抗白血病的免疫应答
王硕1*,范红霞2*,李杨3,郑华国3,李鑫3,李长菲3,陈立钊3,鞠莹3,孟颂东3,4
1 河北大学生命科学学院,河北保定 071002 2 天津医科大学基础医学院,天津 300070 3 中国科学院微生物研究所中国科学院病原微生物与免疫学重点实验室,北京 100101 4 中国科学院大学生命科学学院,北京 100049

王硕, 范红霞, 李杨, 等. CD133表位联合热休克蛋白佐剂疫苗引发抗白血病的免疫应答. 生物工程学报, 2017, 33(6): 1006−1017.Wang S, Fan HX, Li Y, et al. CD133 epitope vaccine with gp96 as adjuvant elicits anantitumor T cell response against leukemia. Chin J Biotech, 2017, 33(6): 1006−1017.
肿瘤干细胞具有肿瘤形成、自我更新和抗化疗的特性,因而受到广泛关注和研究。CD133作为重要的肿瘤干细胞标志物与肿瘤细胞的干性与恶性密切相关。本研究筛选并且鉴定了CD133的3个H2-Kd限制性的细胞毒性T细胞 (CTL) 的表位,分别是CD133419–428、CD133702–710和CD133760–769。将重组热休克蛋白gp96为佐剂联合CD133表位制备表位疫苗,将疫苗免疫CD133+白血病移植的小鼠后引发抗肿瘤的特异性T细胞免疫应答。转输CD133表位特异的T细胞同样可以抑制小鼠淋巴瘤的生长。该研究为设计抗CD133+的白血病和其他肿瘤的表位疫苗提供了依据。
CD133,表位,gp96,白血病,细胞毒性T细胞
10.13345/j.cjb.160481
December 15, 2016; Accepted: March 6, 2017
Supported by:National Basic Research Program of China (973 Program) (No. 2014CB542602), National Natural Science Foundation of China (Nos. 31230026, 81321063, 81471960, 81402840, 81672815).
* These authors contributed equally to this work.
Songdong Meng. Tel: +86-10-64807350; E-mail: mengsd@im.ac.cn
国家重点基础研究发展计划(973计划) (No. 2014CB542602),国家自然科学基金 (Nos. 31230026, 81321063, 81471960, 81402840, 81672815) 资助。
网络出版时间:2017-03-29
http://kns.cnki.net/kcms/detail/11.1998.Q.20170329.1025.001.html
