动态蛋白质网络的构建、分析及应用研究进展
2017-06-23孟祥茂
李 敏 孟祥茂
(中南大学信息科学与工程学院 长沙 410083)
动态蛋白质网络的构建、分析及应用研究进展
李 敏 孟祥茂
(中南大学信息科学与工程学院 长沙 410083)
(limin@mail.csu.edu.cn)
蛋白质组学的快速发展,特别是高通量技术的发展产生了大量的蛋白质相互作用数据,为人们从更深层次理解蛋白质之间的相互作用及其在复杂疾病的作用机理提供了基础.一个生物体内所有的蛋白质与蛋白质之间的相互作用组成的网络称为蛋白质网络.传统的研究多是基于静态的蛋白质网络模型.然而,由于蛋白质自身表达的动态性及蛋白质间相互作用的动态性,真实的蛋白质网络会随着时间和条件不断变化,与疾病的发生和发展有关的蛋白质功能模块也与这种动态变化密切相关.因此,研究者已经把注意力从关注蛋白质网络的静态属性转移到动态属性上,提出了一系列的动态蛋白质网络的构建方法.在介绍静态蛋白质网络的基础上,分类讨论了动态蛋白质网络的构建方法,将现有的动态蛋白质网络的构建方法归纳为基于蛋白质表达动态性的方法、基于多状态下表达及相关性变化的方法和基于时空动态变化的方法这3类:第1类体现的是蛋白质自身表达随时间演化的动态性,第2类则表现为不同条件下蛋白质之间表达相关性的改变,第3类则体现了蛋白质及蛋白质相互作用在时间和空间上的动态变化.然后,对动态蛋白质网络的蛋白质节点和相关子网络进行了动态分析并详细介绍了动态蛋白质网络在复杂疾病中的一些主流应用,如蛋白质复合物识别、蛋白质功能预测、生物标志物识别、疾病基因预测等.最后,对动态蛋白质网络所面临的挑战与未来的研究方向进行了探讨.
蛋白质网络;动态;基因表达;蛋白质复合物;复杂疾病
细胞是由大量的、不同性质的分子通过高度复杂的机制协调作用,从而完成自我复制及对外部扰动的适应等生物过程.蛋白质(protein)是细胞中关键的功能实体,是构成一切细胞和组织结构必不可少的成分,它是生理功能的执行者,也是生命现象的直接体现者[1].随着人类基因组等大量生物体全基因组序列的破译和功能基因组研究的展开,生命科学家越来越关注如何用基因组研究的模式开展蛋白质组学的研究.蛋白质组学(proteomics)指在一个特定细胞或组织或个体中全部蛋白质表达图谱,研究的内容包含蛋白质结构、功能以及相互作用.细胞的生理过程和生命活动(如DNA的复制、基因的调控表达、细胞信号的传导、新陈代谢、细胞增殖与凋亡等)一般都是由多个蛋白质在特定条件下通过复杂的相互作用来实现的[2].在不同周期或者条件下,一个蛋白质可以和不同的蛋白质发生相互作用(protein-protein interaction, PPI)或者同时参与到不同的生命活动中.蛋白质组学的快速发展,特别是高通量技术的发展,使得人们从网络水平深入理解蛋白质的功能、相互作用及其在复杂疾病机理中的作用成为可能.
网络科学理论[3-5]的快速发展为探索复杂的生物体系统提供了新的研究方式.研究者将复杂的生物体系统抽象为生物分子网络,通过构建网络并分析网络的成分关系和网络特性,进而达到对生物体系统深入理解的目的.一个生物体内所有的蛋白质及蛋白质之间的相互作用组成的网络称为蛋白质网络(protein-protein interaction network, PIN),它是我们了解生命活动规律、诠释生命奥秘的基础.基于蛋白质网络,研究人员已经提出了许多方法来挖掘蛋白质组学数据中的信息,其中包括预测蛋白质的功能、预测和推断蛋白质相互作用、识别关键蛋白质、挖掘蛋白质复合物和功能模块、寻找致病候选基因以及识别复杂疾病的生物标志物等.
目前,大部分关于蛋白质网络的研究是基于静态网络模型.然而,静态蛋白质网络是高度平均和理想化的网络结构,包含了在不同条件、不同时间、甚至不同空间发生的各种相互作用.而真实情况是随着外界条件的改变,某些蛋白质会被降解,另一些蛋白质会被翻译出来,由此造成一些蛋白质相互作用的消失和新的蛋白质相互作用形成.蛋白质之间只有在特性条件下,在相同的时间相同的细胞位置才能发生相互作用来完成某种生物过程.显然,传统的静态蛋白质网络没有办法体现这种动态性.如何利用获得的蛋白质相关的生物信息构建和分析动态的蛋白质网络模型是当今生物信息学和系统生物学领域一个很有挑战性的工作.
基因表达有条件和时序地打开和关闭,基因表达数据在生物过程的不同条件或不同阶段能反映蛋白质存在的动态性[6].特别是基因微阵列技术、新一代测序技术产生了海量基因表达数据,为研究不同周期和不同条件下不同的细胞类型的基因表达建立了基础.另外高通量技术的发展,使得产生的蛋白质亚细胞定位数据、质谱数据等其他数据从原来的低通量变为高通量.在多元数据的推动下,动态蛋白质网络的构建成为可能.
本文对现有的动态蛋白质网络的构建方法进行了详细的分类总结,介绍了动态蛋白质网络中蛋白质节点以及相关子网的动态分析的研究进展,并详细介绍了动态蛋白质网络在复杂疾病中的一些主流应用,最后对动态蛋白质网络所面临的挑战与未来的研究方向进行了探讨.
1 静态蛋白质网络
1.1 蛋白质相互作用
蛋白质之间的相互作用根据其作用方式的不同,可以分为物理相互作用(physical interaction)和遗传相互作用(genetic interaction).物理相互作用是指2个蛋白质物理上相互绑定在一起,是一种直接的相互作用关系;而遗传相互作用是在物理上没有发生相互作用,更多地体现在基因之间的功能关联,是一种间接的相互作用关系.通常情况下蛋白质相互作用主要是前者类型.
目前,大规模蛋白质相互作用获取的方式主要有3种:1)高通量实验筛选技术;2)生物信息学计算方法;3)文献挖掘技术.获取蛋白质相互作用的高通量实验方法主要有酵母双杂交技术(yeast two hybrid, Y2H)[7]、串联亲和纯化-质谱分析技术(tandem affinity purification-mass spectrometry, TAP-MS)[8]、蛋白质芯片技术(protein chip technique)[9]等.酵母双杂交技术可以快速、直接分析已知蛋白质之间的相互作用,分离新的与已知蛋白质相互作用的配体及其编码基因.酵母双杂交技术具有高度的敏感性,能够检测到瞬时或较弱的蛋白质相互作用,但它仅能分析细胞核内的蛋白质间的相互作用[10],同时具有较高的假阳性(被检测到的蛋白质相互作用数据在实际中并不存在)和假阴性(潜在的未被检测到蛋白质相互作用数据).串联亲和纯化利用2个亲和标签不同时序来纯化蛋白组件,它能够在真实的生理条件下研究蛋白质的相互作用,同时结合质谱技术的自动化特性,使得大规模地分析相互作用的蛋白质在技术上成为可能[11].相比于酵母双杂交技术,串联亲和纯化质谱分析技术降低了数据的假阳性和假阴性水平[12].蛋白质芯片技术是一种强有力的蛋白质组学研究的新方法,能够进行高通量的蛋白功能分析,它具有特异性、敏感性高等特性,可有效减少药物研发周期并提高医疗诊断效率[13].
生物学家通过这些技术能够方便、大规模地验证蛋白质间的相互作用信息,但是这类技术往往成本费用较高.由于受实验条件等诸多因素的限制,导致不同类型的高通量实验技术以及来自不同实验室相同的高通量实验技术产生了相互作用重叠率较低等问题[14].
相比于高通量实验的方法,利用计算方法预测蛋白质相互作用数据具有周期短、开销少等优势.它综合利用数学、物理和信息学等多学科的理论知识,通过计算机建模来预测未知的蛋白质相互作用数据.基于计算预测的方法可以归纳为5种[15-16]:
1) 基因组信息关联推断;
2) 基于遗传进化关系的方法;
3) 基于蛋白质一级序列信息推断;
4) 基于蛋白质三维结构信息的方法;
5) 蛋白质网络分析法.
由于计算预测方法采用不同的蛋白质相互作用数据库,往往预测结果也有较大的差异性,目前还没有比较好的统一评价标准.
基于文献挖掘的蛋白质相互作用获取方式主要是依赖于海量的关于蛋白质相互作用的文献报道.这些文献不仅包括了蛋白质相互作用数据信息,有的还给出了实验的条件、环境及蛋白质注释、亚细胞定位等多种信息,并且这些信息还在不断地增加与更新.丰富的文献信息为挖掘蛋白质相互作用数据提供了基础,目前主要采用基于自然语言处理(natural language processing, NLP)的文本挖掘技术[17-18].文献挖掘的蛋白质相互作用数据信息得到了生物学实验的支撑,可靠性较高,同时也能为高通量实验筛选技术和计算预测得到的蛋白质相互作用数据提供文献参考依据.但受限于科学文本的复杂性和人类语言表述的多样性,如何有效地提取文献中的蛋白质相互作用数据信息仍然是很有挑战的工作.
目前,已经产生了大量可用的蛋白质相互作用数据,而且这些数据还在持续不断地增加.研究者已经成功地构建了多个不同的蛋白质相互作用数据库,为蛋白质组学的相关研究提供了海量的数据来源.Pathguide网站*http://pathguide.org提供了丰富的蛋白质相互作用数据库的相关信息,本文选取部分常用的蛋白质相互作用数据库,如表1所示:
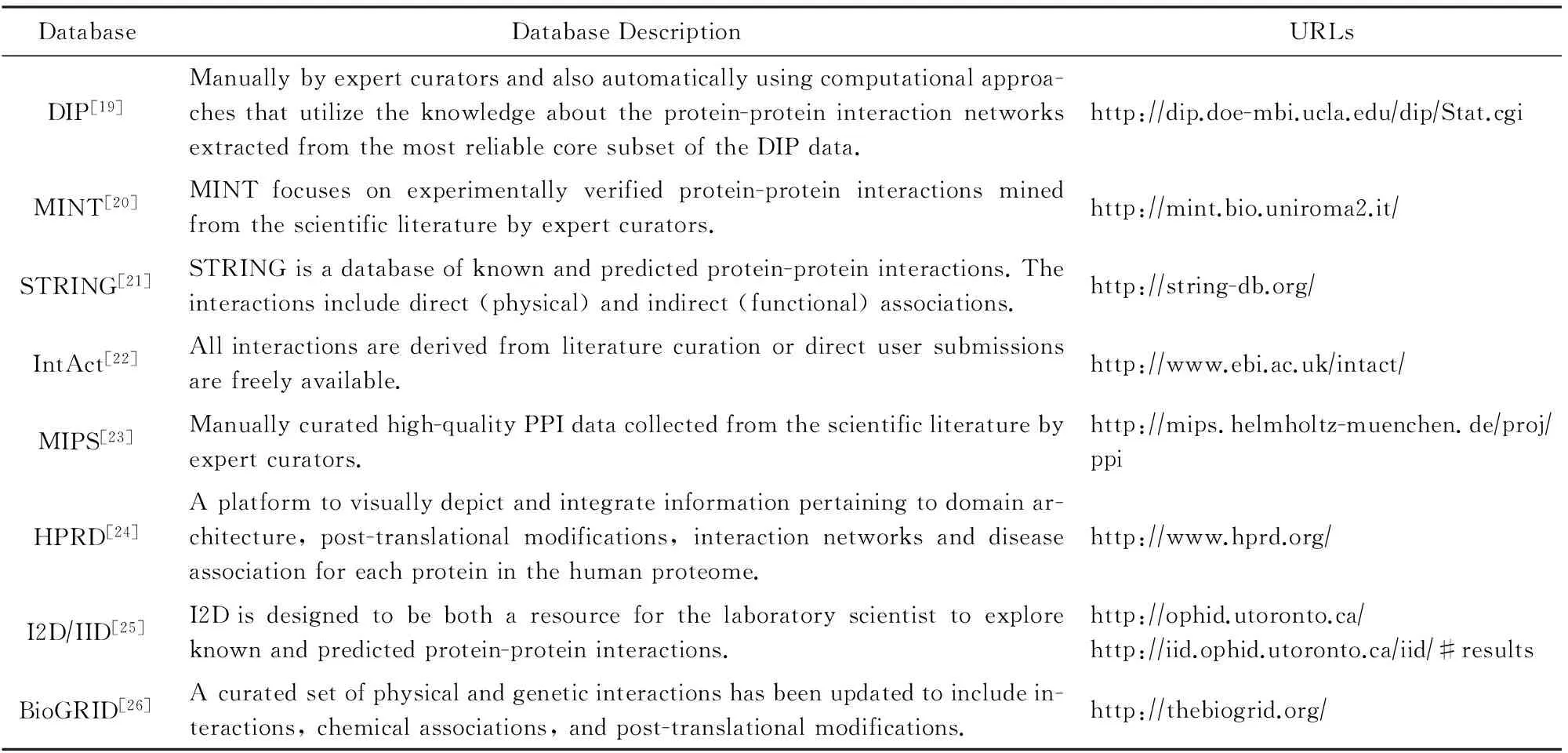
Table 1 The Commonly Used PPI Databases表1 部分常用的蛋白质相互作用数据库
1.2 静态蛋白质网络
通常情况下,静态蛋白质网络可以用一个无向图G=(V,E)表示,其中V表示图中节点集合,E表示图中边的集合.映射到蛋白质网络中,每个节点就是一个蛋白质,每条边则是蛋白质之间的相互作用.根据边取值的差异,无向图又可分为加权图和非加权图.非加权图中的每条边权重相同,反映蛋白质之间的相互作用视为同等地位,通常简单地用二进制值0和1表示2个蛋白质间是否存在相互作用关系(1表示存在,0表示不存在).加权图中的每条边权重不同,说明蛋白质之间的相互作用具有差异性.
对于蛋白质网络早期的研究,研究者重点分析了网络的拓扑特性,如连接度、网络直径、中心性和集聚系数等[27-30].同时,这些网络分析发现蛋白质网络具有无标度特性[31-33]、小世界性质[34-36]、功能模块化结构[37-39]等.无标度特性表现为蛋白质节点的度服从幂律分布,即蛋白质网络中大部分蛋白质只有少量邻居相连,而个别蛋白质却有众多的邻居节点相连接.这些有大量邻居节点的蛋白质称为hub节点,这些hub节点影响着整个蛋白质网络,对于细胞生存至关重要.利用这种特性,研究者可以根据蛋白质在网络中的拓扑中心性来识别关键蛋白质[40-44].而小世界特性和功能模块结构则表现为网络具有较高的集聚系数,往往形成模块化结构,这些模块通常对应于蛋白质复合物或者功能模块.一般情况下,生命活动的发生与发展都需要多个蛋白质共同协作形成的大分子蛋白质复合物或者功能模块来完成.这些蛋白质复合物或者功能模块中有一部分是稳定的,参与生命周期中的多个生理过程,还有一部分是临时形成、动态的.蛋白质在不同的外在条件或刺激下,具有不同的功能.生物信息学中常用图聚类的方法,识别蛋白质网络中的一组相互作用且具有一定功能的子图作为蛋白质复合物或者功能模块(一般都称为cluster)[45-48].目前主要的蛋白质网络图聚类方法可分为识别稠密子图的聚类方法、层次化的聚类方法以及融合多元信息的聚类方法等[46].大部分蛋白质网络分析的图聚类算法都是基于无向图模型的,其中有些方法是基于非加权图的,有些方法是基于加权图的,还有些方法既可以用于加权图又可用于非加权图.
基于蛋白质网络,另一个重要的研究方向是网络比对问题[49-50]和子网络查询问题[51-52].蛋白质网络的比对主要是依据网络的相似性计算,从待比较的2个或多个蛋白质网络中找出保守的子网,进一步预测蛋白质复合物以及特定功能的通路,预测新的蛋白质相互作用以及不同物种间的保守进化关系等[53-55].蛋白质子网络查询目标是从一个大规模蛋白质网络中识别出一个与给定的查询网络高度相似的子网区域[56].针对查询模式的比对问题,研究者们已开展了大量的研究工作,提出了很多有效的解决方法[57-58].
目前,基于静态蛋白质网络的研究已取得了较大的进展,但在实际的生物系统中分子网络是时刻在变化的,因此构建动态蛋白质网络才能模拟真实的生物系统的运行规律.接下来本文将对现有的一系列动态蛋白质网络的构建方法进行系统的分类总结.
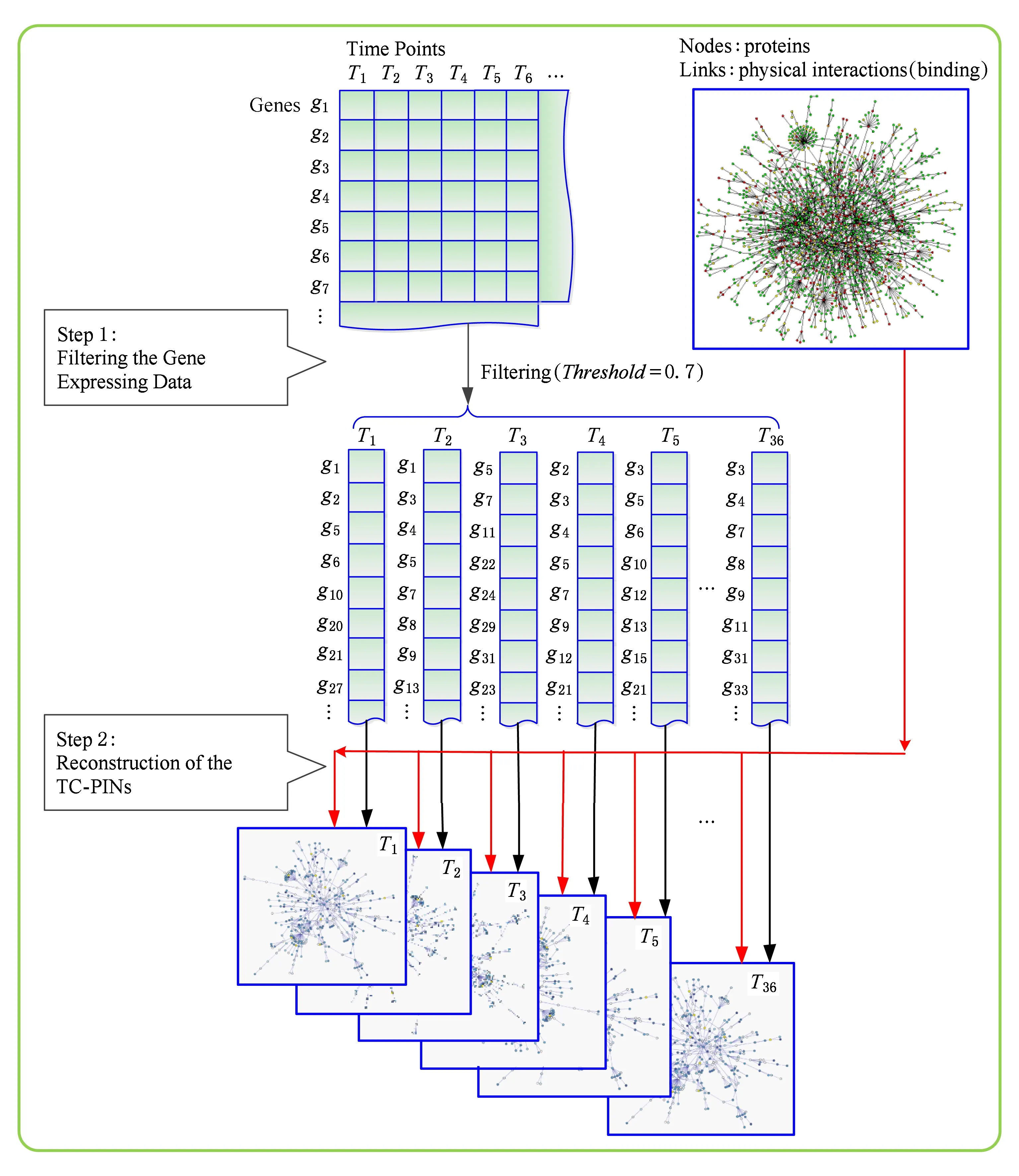
Fig. 1 Schematic diagram of the time course dynamic PIN construction图1 一种时序动态蛋白质网络构建示意图[60]
2 动态蛋白质网络的构建方法
动态蛋白质网络可以用一个无向图集合G={G1,G2,…,Gi,…,Gn}表示,其中Gi=(Vi,Ei)表示第i时刻或条件下的子网,Vi表示图中第i时刻或条件下的节点集合,Ei表示图中第i时刻或条件下的边集合.蛋白质和蛋白质间的相互作用都会随着外界刺激或条件改变而变化,因此由蛋白质和蛋白质间的相互作用构成的蛋白质网络也应该是受到外界环境条件约束的,是一个时刻在动态发展的.根据蛋白质网络所包含组件(蛋白质节点和蛋白质间的相互作用边)属性的动态性,本文把现有的动态蛋白质网络的构建方法归纳为3类:基于蛋白质表达动态性的方法、基于多状态下表达及相关性变化的方法和基于时空动态变化的方法.
2.1 基于蛋白质表达动态性的方法
蛋白质的表达具有时空动态的特性:从时间维度上分析,蛋白质表达动态性主要表现为蛋白质在某些特定的时刻表达,而在其他时刻没有表达;从空间维度上来看,蛋白质的表达动态性体现在蛋白质只在某些特定的组织中表达,而在其他组织中不表达.因此,如果在一个细胞中或者某个时刻,2个蛋白质都不表达,那么它们之间的相互作用就不会发生[59].静态的蛋白质网络能够提供细胞内蛋白质间的相互作用行为的定性描述,为动态蛋白质网络的构建提供了一个的支架;而基因表达数据能够反映出不同时刻/组织/条件下细胞中被转录的mRNA定量信息.由中心性法则可知,蛋白质是由mRNA翻译而来,从而可以从基因表达信息中获得蛋白质表达的动态性信息.有效结合这2种定性和定量信息,能够阐述细胞内蛋白质之间的动态组织形式.基于蛋白质表达动态性的方法的基本思路是:1)根据基因的表达信息判断蛋白质在各个时刻/组织/条件下表达或未表达;2)结合静态蛋白质网络,构建每个时刻/组织/条件的蛋白质子网;3)由这些反映蛋白质表达动态性的子网构成了动态变化的蛋白质网络.其中每个时刻/组织/条件的蛋白质子网由这个时刻/组织/条件下表达的蛋白质及其相互作用构成.图1给出了一种时序动态蛋白质网络构建的示意图.
由于微阵列技术或新一代测序技术产生的高通量基因表达数据存在不可避免的背景噪音,因此这类构建方法的关键在于如何判断蛋白质在各个时刻/条件下/组织中的表达动态性.在已有的动态蛋白质网络构建中,判断蛋白质表达动态性的方法不尽相同,表2给出了6个主要的基于阈值的动态蛋白质网络构建方法.本文把基于阈值的方法分为2类:固定阈值(fixed threshold)和动态阈值(active threshold).
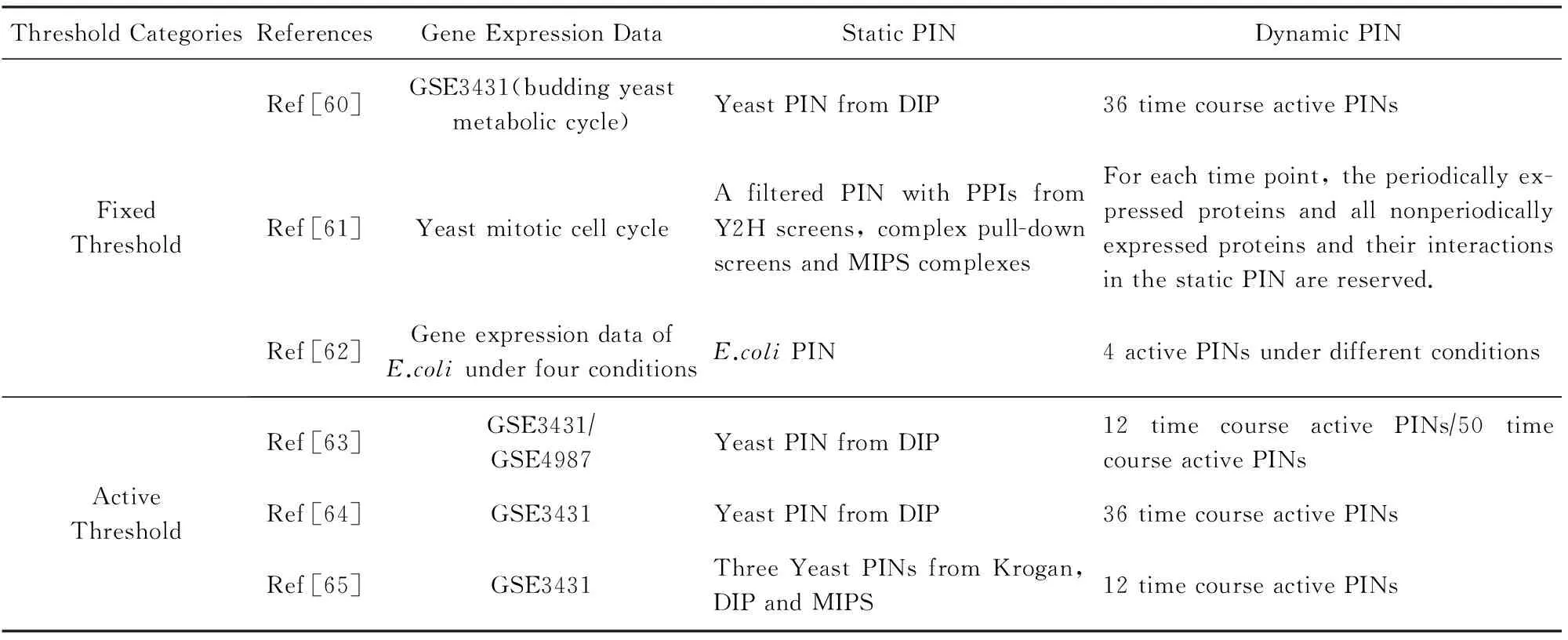
Table 2 Dynamic PIN Construction Methods Based on Protein Presence Dynamics表2 基于蛋白质表达动态性的动态网络构建方法
2005年,De Lichtenberg等人[61]构建了基于时序基因表达数据的酵母动态蛋白质网络.基于时序基因表达数据,蛋白质被分为周期性表达蛋白质和持续性表达蛋白质2类[66],他们认为仅周期性表达的蛋白质存在动态性,且仅在基因表达数据中峰值所对应的时刻表达.对于周期性表达的蛋白质,蛋白质p的活性出现时刻T(p)为
T(p)={i|Max(Exp(p,i)),i=1,2,…,t},
(1)
其中,Exp(p,i)是蛋白质p在时刻i的基因表达值;对于持续性表达的蛋白质,蛋白质活性出现时刻如式(2)所示:
T(p)={i|i=1,2,…,t}.
(2)
因此,每个时刻的动态蛋白质子网由该时刻出现的周期性表达蛋白质和所有持续性表达的蛋白质以及它们之间的相互作用组成.基于预处理得到的小规模高可靠性的蛋白质相互作用以及周期性表达蛋白质识别策略,对比包含5 000多个蛋白质的酵母蛋白质网络,这个酵母动态蛋白质网络只包含300多个蛋白质,丢失了大量的蛋白质及其动态信息.另外,仅将基因表达数据中峰值所对应的时刻作为周期性表达蛋白质的表达时刻不符合生物事实,从而导致大量的蛋白质表达动态性信息的丢失.后续的很多研究者都开始采用阈值的方法来确定蛋白质的表达动态性.Hegde等人[62]构建了大肠杆菌在4个不同条件下的蛋白质网络来研究不同条件下蛋白质相互作用的动态改变.他们将基因芯片中每一个区域的平均表达水平作为该区域的阈值来区别噪音值和真实表达值.对于某个条件i下的某个j扇区s(i,j),其包含基因数记为ns(i,j),则其活性阈值θ(s(i,j))为
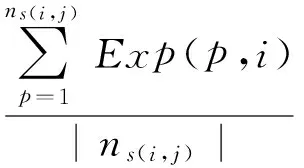
(3)
蛋白质活性出现时刻为
T(p)={i|Exp(p,i)≥θ(s(i,j)),i=1,2,…,t}.
(4)
2011年,Tang等人[60]基于大量周期性表达的基因在酵母代谢周期中表达峰值都会大于一个常量这一现象[67],采用一个固定阈值来判断蛋白质的表达动态性,并基于此构建了酵母的时序动态蛋白质网络,其蛋白质活性出现的时刻计算为
T(p)={i|Exp(p,i)≥θ,i=1,2,…,t},
(5)
其中,阈值θ是一个常量.这种固定阈值的选取依赖于对酵母周期性表达基因在某个具体的基因表达数据中峰值分布的研究,因此很难应用在同一物种的其他基因表达数据以及其他物种的基因表达数据上.另外,许多在酵母细胞周期转录水平一直很低的mRNA很容易被这个固定阈值过滤掉,而实际上这些mRNA也可能会被翻译成蛋白质[68],这会使得构建的动态蛋白质网络不可避免地丢失一些蛋白质以及它们的动态表达信息.从这些问题出发,考虑到不可避免的背景噪音和每个基因各自的表达特性,Wang等人[63]提出了一个基于3-sigma法则的方法来根据每个基因的表达曲线为基因对应的蛋白质设计一个阈值,用于判断该蛋白质在什么时刻表达并处于活性状态.活性阈值由式(6)求出:
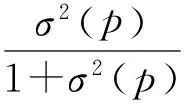
(6)
其中,μ(p)和σ(p)是蛋白质p的基因表达的算术平均值及标准差;蛋白质活性出现时刻如式(7)所示:
T(p)={i|Exp(p,i)≥θ(p),i=1,2,…,t}.
(7)
基于识别的每个蛋白质活性表达时刻点,构建了动态蛋白质网络.每个时刻上的动态蛋白质子网由该时刻的处于活性的蛋白质及其相互作用组成.Xiao等人[69]则采用一个动态模型方法,将基因表达数据分为时间相关的和时间不相关的2类.和时间相关的基因表达数据更有可能是动态表示的,而不是随机的;而和时间不相关的基因表达数据则更有可能是随机性的.如果基因是时间独立性的,并且它们的平均值是非常小的,那么这些基因表达数据就被认为是噪声,通过这种动态模型来过滤基因表达数据中的无效数据.同时在3-sigma方法的基础上,设计了新的阈值函数来计算确定过滤了噪声后的基因(蛋白质)活性时间点,从而构建酵母的动态蛋白质网络.而Shen等人[64]认为3-sigma方法虽然能够取得相对较好的结果,但是会过滤掉一些一直有较高表达信息的蛋白质,造成有用数据的丢失.在3-sigma基础上,他们提出了偏差度(deviation degree)的方法来判断蛋白质的活性时刻,进而构建时间演化的动态蛋白质网络,其活性阈值为
θ(p)=μ(p)+σ(p).
(8)
蛋白质活性出现时刻为
T(p)={i|Exp(p,i)≥θ(p),i=1,2,…,t}.
(9)
由于蛋白质间相互作用强度的异质性,定量描述动态蛋白质网络中的相互作用强度对于真实反映细胞进程的作用机制有重要作用.他们采用连接亲密度来量化蛋白质之间相互作用的强度,构建了加权的动态蛋白质网络,从而降低了假阳性数据以及假阴性数据(瞬时PPI数据的丢失)对网络可靠性的影响.Zhang等人[65]认为简单的描述蛋白质是表达状态或未被表达状态不符合实际蛋白质表达的过程,而应该是蛋白质在不同的时刻组织条件下表现为具有不同水平的表达活性.基于3-sigma方法,他们设计了k-sigma(k=1,2,3)的阈值方法.当k取不同数值时,判断蛋白质是否表达的活性阈值也随之变化.当蛋白质实际的表达量处于不同的阈值区间,则其对应的处于活性状态的概率值也不同.基于蛋白质在不同时刻的活性概率,他们构建了基于概率的动态蛋白质网络,其活性阈值为
(10)
其中,k=1,2,3.当k=1时,活性阈值θk(p)记为θ1(p);当k=2时,活性阈值θk(p)记为θ2(p);当k=3时,活性阈值θk(p)记为θ3(p).基于3-sigma法则,Zhang等人[65]将蛋白质p在时刻i的活性概率Proi(p)分为4个等级,具体计算为
(11)
蛋白质p的活性概率表示其在某时刻的活跃水平,可以根据活性概率判定p在该时刻是否是活性的.
目前,基于表达动态性的蛋白质网络的构建方法主要采用阈值来区分基因表达数据中噪音和真实表达,从而提取蛋白质的表达动态性信息.然而,大多数阈值方法缺少对噪音的系统性分析和必要的理论支持,使得在不同的基因表达数据上有较大的应用局限性.
2.2 基于多状态下表达及相关性变化的方法
蛋白质表达水平的改变可能导致蛋白质之间相互作用强度的增加或减少,也会引起蛋白质之间表达相关性的改变,从而导致生理状态的改变.表达方差(EV)可以用来衡量蛋白质的动态性,具有小EV值的蛋白质的动态性很低,反之,蛋白质的动态性较高.利用相关性计算方法(例如皮尔逊相关系数PCC)可以衡量一对蛋白质的表达相关性,表达相关性越高,它们在细胞中同时表达的机会越多,更容易发生相互作用;反之,表达相关性越小,它们在细胞中同时表达的机会越少,那么它们发生相互作用的可能性就越小.目前主要的基于多状态下表达及相关性变化的动态蛋白质网络构建方法如表3所示:
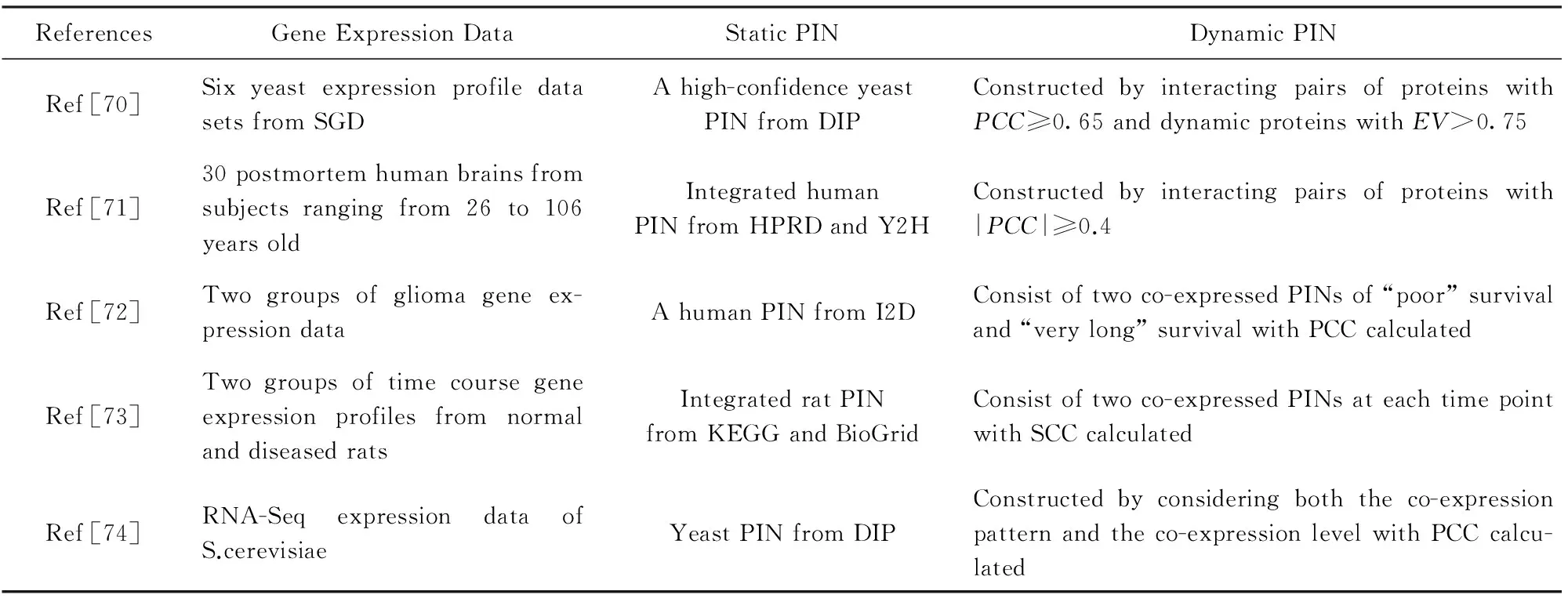
Table 3 Dynamic PIN Construction Methods Based on Multiple States Expression and Correlation Alteration表3 基于多状态下表达及相关性变化的动态蛋白质网络构建方法
基于包含多种状态的基因表达数据,通过计算表达相关性,可以研究同种细胞不同状态下的蛋白质网络的动态性.例如Komurov等人[70]在6个不同数据集上的272个基因表达数据上计算各个蛋白质的EV,并基于EV将蛋白质分为动态蛋白质(EV>0.75)和静态蛋白质(EV<0.25)两类.他们提出动态蛋白质更趋向于处在一个高度表达相关的环境中,从而构建了一个由PCC计算的表达相关性大于或等于0.65的相互作用蛋白质对组成的简单的动态蛋白质网络,其中动态蛋白质占绝大多数.为研究人类蛋白质网络的动态特性,Xia等人[71]基于26~106岁年龄阶段的30个人的大脑基因表达数据建立了一个由正相关(PCC>0.4)和负相关(PCC<-0.4)的相互作用蛋白质对组成的动态蛋白质网络.这个动态网络包含了衰老过程中表达相关性较高的蛋白质以及它们之间的蛋白质相互作用.在Xia等人研究的基础上,Xue等人[75]构建了类似的人类和果蝇的动态蛋白质网络,用来研究在人类和果蝇衰老过程中蛋白质网络结构的动态变化.在这些动态网络中,大多数相关性高的相互作用的蛋白质拥有较高的EV值,但是并非所有的动态蛋白质都包含在这种动态网络中.另外,在这种动态网络中,包含的只是在多状态下表达相关性高的蛋白质之间的相互作用,这些相互作用不一定都是同时发生的.因为表达相关性是通过蛋白质的表达来间接刻画它们之间相互作用会发生的可能性,而这些相互作用到底什么时候发生就不得而知了.
近年来,一些研究者开始关注基于表达相关性的动态蛋白质网络构建,特别是不同病理状态下的动态蛋白质网络.例如Zhang等人[72]基于2组不同发展状态下的神经胶质瘤病人的基因表达数据(短存活的样本和长存活的样本),计算了与胶质瘤相关的蛋白质分别在2组不同发展状态下的表达相关性,构建了胶质瘤动态蛋白质网络,其动态性主要体现为表达相关性的变化及表达信息的变化.类似于Zhang等人的构建方法,为了研究二型糖尿病,Sun等人[73]利用2组正常小鼠和疾病小鼠在3个二型糖尿病相关组织上的时序基因表达数据以及小鼠的蛋白质网络,分别计算每个组织在每个时刻点上相互作用的蛋白质的表达相关性,识别出各个时刻点上显著性差异表达的蛋白质以及相关性具有显著性差异的相互作用,构建了差异性的动态蛋白质子网.与Zhang等人不同的是,Sun等人采用了斯皮尔曼相关系数(SCC)来计算2个相互作用的蛋白质的表达相关性,并且动态蛋白质网络不仅包含了在相关性上具有显著性差异的相互作用和具有显著性表达差异的蛋白质,还包含了显著性表达差异的蛋白质之间的相互作用.Shang等人[74]采用RNA-seq数据代替基因表达数据来构建动态蛋白质网络,他们认为相互作用的蛋白质对具有一致的表达模式.通过基因的共表达模式过滤具有一定噪声的相互作用,然后再选取表达相关性PCC≥ 0.95的相互作用蛋白质对构建动态的蛋白质网络.RNA-seq数据背景噪声更小,同时还能够检测未知的转录物和亚型[76].因此,利用RNA-seq数据构建动态蛋白质网络是今后的重要研究方向.

Fig. 2 Overview of the paradigm for constructing the spatial and temporal active PIN图2 一种时空动态蛋白质网络构建示意图[84]
在上述基于多状态下表达及相关性变化的动态蛋白质网络的构建中,不同的相关性计算方法和差异显著性分析方法影响动态网络的规模,对动态蛋白质网络的构建非常关键.不同样本下的不同背景噪音不可避免地会对相关性计算以及显著性分析造成影响.不同的物种,显著性差异判断的阈值显然也不同.目前,多状态下的动态蛋白质网络的构建还处于初步研究阶段,只刻画了多状态下的表达及相关性差异.由于缺少新的描述模型和构建方法,不能对各种状态下蛋白质网络的整体变化等进行研究.因此,提出新的精确刻画多状态下蛋白质网络的描述方法非常必要.
2.3 基于时空动态变化的方法
基于蛋白质表达动态性的方法体现的是蛋白质自身表达随时间演化的动态性;基于多状态下表达及相关性变化的方法则表现为不同条件下蛋白质之间表达相关性的改变;而基于时空动态变化的方法则体现了蛋白质及蛋白质相互作用在时间和空间上的动态变化.现阶段动态蛋白质网络构建的研究主要体现了蛋白质在时间动态性上的信息,并且取得了一系列相关研究成果.随着蛋白质组学研究的快速发展,体现蛋白质空间动态性上的信息数据也越来越多.结合不同组织的基因表达数据构造的组织特异性蛋白质网络,能够反映蛋白质在空间上的表达动态性:在不同的组织中,某些蛋白质只能在特定的组织细胞中表达,从而影响相互作用的发生.例如,Bossi等人[77]基于79个不同的人体细胞或组织的基因表达数据[78]和人类蛋白质网络,构建了79个组织特异性的蛋白质网络.他们也利用固定阈值的方法来区分所有基因表达数据中的噪音和真实表达[79].但蛋白质的空间动态性准确来说,是指在不同的细胞生长条件下和应对外部刺激时蛋白质在细胞中的位置会发生变化,从而影响相互作用的发生.例如,当细胞应对DNA损坏时,Boisvert等人[80]观察到蛋白质的位置发生了改变.Zhao等人[81]和胡赛等人[82]结合时序基因表达数据、结构域信息和复合物信息,构建了动态加权蛋白质网络.他们认为蛋白质在某时刻的表达水平超过自身的平均表达,则蛋白质在该时刻表达.不同于3-sigma方法,他们认为取消了阈值能够提高算法的适应性.
根据蛋白质受到时间和空间的约束,蛋白质之间只有在相同时间和相同细胞位置才能发生相互作用[83],我们构建了具有时空特性的动态蛋白质网络[84],该网络具体的构建流程如图2所示.首先,我们采用改进的3-sigma动态阈值方法判断时序基因表达数据中蛋白质活性信息;然后利用蛋白质亚细胞定位数据获得蛋白质在细胞中的位置信息;最后结合发生在相同时间相同位置的相互作用信息,构建成时空动态蛋白质网络.相比于静态的蛋白质网络,我们构建的时空动态蛋白质网络体现了蛋白质及蛋白质之间相互作用随时间和空间位置变化的特性.
现有的动态蛋白质网络构建方法主要还是蛋白质自身表达水平的动态变化过程,而蛋白质相互作用强度的动态变化过程研究尚处于基础研究阶段.因此,如何同时结合时间、空间动态性信息构建更加有效的动态蛋白质网络还需要投入大量的研究精力.
3 蛋白质节点及相关子网络的动态变化分析
蛋白质以高效的、精确的、特异性的方式来发挥功能,同时蛋白质结构与功能的动态变化使得蛋白质具有很强的适应性和进化性[85].在静态蛋白质网络分析中我们可知,蛋白质网络中存在少量hub类型的蛋白质节点,这些蛋白质连接度较大,往往是在不同时刻或不同条件下形成的相互作用关系.而Han等人[86]则发现酵母动态蛋白质网络中的这个hub类型的蛋白质又可分为party hub和date hub.party hub类型的蛋白质往往处于蛋白质功能模块的中心位置,与多个蛋白质同时发生相互作用且具有较高的共表达水平;而date hub类型的蛋白质则处于功能模块之间,在不同时刻或细胞位置与多个蛋白质发生相互作用且共表达水平相对较低.Taylor等人[87]受此启发将hub类型的蛋白质分为intermodular hub和intramodular hub.他们的研究发现intermodular hub类型的蛋白质表现出较高的集聚系数,intramodular hub类型的蛋白质则表现出较高的介数.
区分蛋白质网络中的动态节点和静态节点,并对网络中动态性最强的节点及属性进行分析有助于理解蛋白质的功能和蛋白质网络的组织结构[88].例如,在不同条件下,考虑网络中基因表达水平的变化,可以筛选出一些差异性表达的基因,这些基因往往跟疾病的发生与发展密切相关.
在细胞中,实际的蛋白质之间的相互作用也不是一成不变的,而是随着时间和条件不断变化的,这就体现了蛋白质的相互作用动态性[89-90].而由蛋白质和蛋白质之间的相互作用构建的蛋白质子网也具有动态性,在不同条件下所构建的蛋白质子网称为条件特异性子网,如蛋白质复合物/功能模块、组织特异性子网、内容相关子网等[88].从不同层面动态分析条件特性子网有利于整体或局部理解蛋白质网络的组织结构和生物体系统功能机制[91-92].传统研究都是从全局角度去识别静态的蛋白质复合物或功能模块,并且难以精确区分二者.而实际的蛋白质复合物更多的是动态单元,特别是一些瞬时形成的蛋白质复合物在静态蛋白质网络中难以检测到.部分基因只能在特定的细胞组织中表达,只有2个基因在相同的组织中,它们才能够发生相互作用.基于组织特异性子网能够发现组织特异性蛋白质与广泛表达蛋白质具有显著性的差别[77,93-94].而内容相关子网的研究重点关注的是如何识别出特定复杂疾病的生物标志物或者筛选出疾病组和对照组差异性的基因集合,这对于疾病的早期诊断、药物设计和临床应用具有很强的实用性[95-96].常用基于图搜索的内容相关子网识别方法[97-99],主要分2个步骤实现:1)定义打分函数来量化不同条件下网络结构的改变程度;2)设计搜索算法提取得分最高的条件特异性子网[88].
4 蛋白质网络的动态模拟及可视化
网络的动态模拟可以反映出生物细胞对于外界刺激时的响应过程,以及分子网络随时间的演化过程.蛋白质网络的动态模拟可以呈现蛋白质及蛋白质之间的相互作用在细胞中所参与的生命活动过程规律,有助于理解蛋白质复合物/功能模块的形成机制以及差异性表达基因及异常通路在疾病病变中所扮演的角色[100-101].基于计算的方法为蛋白质网络的动态模拟提供了有效手段.常用的模拟分子网络动态的数学模型有布尔模型、逻辑模型、贝叶斯模型和微分方程模型等[88],这些模型同样适用于蛋白质网络的动态建模分析.小规模蛋白质网络,可采用微分方程模型来进行动态模拟;而对于大规模的蛋白质网络则需借助不需要反应参数的不太精确的模型,如智能建模[88,102].
网络可视化技术作为一种重要的辅助手段,可以帮助研究人员直观地观察网络的结构并有助于进一步挖掘隐藏的信息.Cytoscape[103]是一个十分重要开源的支持插件开发的软件系统,可用于大规模蛋白质相互作用等复杂生物网络的可视化分析.由于Cytoscape界面友好、功能齐全、数据库整合较好,同时还可以通过简易插件对其功能进行扩展,Cytoscape软件包的功能日益丰富和完善.基于Cytoscape的蛋白质网络分析的插件有很多,例如分析节点拓扑特性的插件CytoNCA*http://apps.cytoscap-e.org/apps/cytonca[104],以及用于网络聚类分析的插件MCODE*http://apps.cytoscape.org/apps/mcode[105],ClusterViz*http://apps.cytos-cape.org/apps/clusterviz[106]、CytoCluster*http://apps.cytoscape.org/apps/cytocluster,Cluster-ONE*http://apps.cytoscape.org/apps/clusterone[107],clusterMaker2*http://apps.cytoscape.o-rg/apps/clustermaker2等.基于Cytoscape的动态蛋白质网络可视化插件有DyNet*http://apps.cytoscape.org/apps/dynet[108],DynNetwork*http://apps.cytoscape.org/apps/dy-nnetwork.DyNet是一个功能十分强大的动态网络分析工具,能够对多状态的动态分子网络提供实时同步的动态可视化分析,能够识别出不同网络状态下的最大保守子网模块.DynNetwork提供了多种网络中心性测量指标,比如度中心性(degree centrality)[109]、接近中心性(closeness centrality)[110]、介数中心性(betweenness centrality)[111]等.
此外,其他一些复杂网络的可视化分析工具也可以对动态网络的子网进一步分析.Gephi[112]可用于动态和分层图的交互可视化与探测.Osprey[113]使用不同颜色标识基因的功能和相互作用数据,还可以让用户通过基因名称进行文本搜索和它相关的蛋白质等功能信息.Pajek[114]是大型复杂网络分析工具,不仅实现了一整套快速有效的用来分析复杂网络的算法,而且提供了一个用于可视化分析的界面.Pajek不仅可以对普通网络进行可视化,还支持多关系网络的可视化呈现.C-DEVA[115]基于Java的综合生物网络分析平台,集成了多种算法应用于生物网络功能模块的预测、评估、可视化以及功能富集分析等,且具有很好的扩展性.
5 动态蛋白质网络在复杂疾病中的应用研究
动态蛋白质网络构建方法的发展,有效克服了基于静态蛋白质网络分析方法的局限,在蛋白复合物/功能模块识别、生物标志物识别、疾病相关研究方面都起到了很好的作用.另外,复杂疾病药物研究也开始由标靶某个蛋白质或基因转向系统地标靶蛋白质及蛋白质间的相互作用构成的子网.
5.1 蛋白质复合物识别
蛋白质复合物和网络功能模块都是一组相互作用的蛋白质集合,它们的区别在于蛋白质复合物中的蛋白质是在同一时间同一地点发生的相互作用,而网络功能模块中的蛋白质是在不同时间不同地点(如细胞不同阶段或条件、不同细胞位置等)发生相互作用来完成某一生物进程.静态的蛋白质网络没有涉及时间和空间的信息,不能够对蛋白质复合物和功能模块精确区分.另外,静态的蛋白质网络带来的噪声也会影响算法检测结果的准确性.动态的蛋白质网络体现了真实的蛋白质复合物/功能模块形成过程中体现的动态性,在动态蛋白质网络上进行蛋白质复合物/功能模块识别具有重大的优势.
De Lichtenberg等人[61]在他们所构建的动态蛋白质网络的上下文中,对MIPS数据库[23]的已知蛋白质复合物进行分析,发现大部分复合物由周期性表达和持续性表达的蛋白质构成,并且这些复合物的形成机制符合一种即时组合机制(just-in-time).Tang等人[60]和Wang等人[63]分别在动态蛋白质网络上预测蛋白质复合物,发现在动态蛋白质网络上预测的蛋白质复合物更加准确、更加具有功能一致性.在人类蛋白质网络中,Calvano等人[116]研究人体血液白细胞在不同时间点对内毒素的反应,且鉴别出被次内毒素刺激扰乱的重要蛋白质功能模块.Luo等人[117]提出了一个基于条件的共调控蛋白复合物识别框架,并在细胞周期、DNA损伤等条件下对酵母数据集进行了测试.为了研究动态蛋白质网络的模块化结构,Xia等人[71]在只包含正相关或负相关的蛋白质对的动态蛋白质网络中,运用层次聚类的方法识别蛋白质复合物,发现了转录反相关模块是一种细胞状态切换的开关.在有关衰老过程的研究中,Xue等人[75]分析了果蝇、人类在动态蛋白质网络中的模块化结构,发现只有小部分蛋白质功能模块在表达中的变化与老化有关.Ou-Yang等人[118]利用3-sigma方法判断每个蛋白质的活性,同时根据PCC阈值把相互作用分为瞬态相互作用(动态部分)和稳态相互作用(静态部分)来构建动态蛋白质网络.结果发现在构建的动态蛋白质网络上预测的重叠的动态蛋白质复合物更能体现真实的蛋白质复合物及其动态特征.Shen等人[64]在其构建的动态蛋白质网络上,根据连接亲密度的思想,进行动态蛋白质复合物的预测,找到具有生物意义功能的蛋白质复合物.Lei等人[119]基于3-sigma方法和改进的MCL聚类算法及优化思想,在构建的动态蛋白质网络上进行蛋白质复合物预测.相比于静态蛋白质网络,动态蛋白质网络能够预测出更加精确的蛋白质复合物,这有助于我们理解蛋白质复合物的形成机制及其在细胞中所发挥的功能作用.
5.2 蛋白质的关键性及功能预测
关键蛋白质是维持生物体生命活动必不可少的生物大分子,没有了它们生物体将不能存活或生长[120-121].关键蛋白质的研究对合成生物学的基础研究、设计新的抗菌药物等具有很重要的帮助.基于网络水平的关键蛋白质预测及其应用研究已经成为生物信息学和系统生物学领域研究的热点方向.目前,已有的大量的关键蛋白质预测方法被提出.如基于拓扑特性的方法有度中心性(degree centrality)[109]、接近中心性(closeness centrality)[110]、介数中心性(bet-weenness centrality)[111]、子图中心性(subgraph centra-lity)[122]、节点的局部平均连通性(local average connec-tivity)[123]、邻居中心性(neighborhood centrality)[124]和集成复合物中心性(united complex centrality)[125]等;还有融合其他生物信息的方法ION[126],SON[127]等.这些方法在一定程度上都取得了较好的效果,但还远远不够,其主要原因是这些方法都是基于静态的蛋白质网络分析的.Xiao等人[128]结合静态蛋白质网络和时序基因表达数据构建了一个时序动态蛋白质网络,并在此网络上进行关键蛋白质预测,结果发现动态蛋白质网络比静态蛋白质网络能够更加有效地预测关键蛋白质.我们基于3-sigma构建了动态蛋白质网络,然后再结合亚细胞定位信息对动态蛋白质网络进行了净化处理,最后在净化的动态蛋白质网络上进行关键蛋白质预测,同样发现动态蛋白质网络比静态蛋白质网络能够更加有效地预测关键蛋白质[129].
基于蛋白质网络预测未知的蛋白质功能是生物信息学中一个十分重要的研究课题.特别是动态蛋白质网络的快速发展,通过融合多元生物信息,能够提供一个更加有效的网络,提高蛋白质功能预测的准确率.Zhao等人[81]认为减少假阳性和假阴性造成的负面影响是提高蛋白质功能预测性能的关键和瓶颈,利用蛋白质结构域信息、蛋白质复合物信息及蛋白质网络拓扑特性构建加权动态蛋白质网络来进行蛋白质功能预测,实验结果验证了动态加权网络的有效性.Greene等人[130]构建了一个包含144个人类组织和细胞类型的组织特异性交互网络,从全基因关联的角度预测蛋白质功能.
5.3 生物标志物识别
生物标志物是一种在正常生理过程、病理或治疗过程中能够客观测量的生物信号,能够对疾病进行早期的诊断并预测和监测治疗反应和不良反应,也是生物体受到损害时的重要预警指标[131-133].传统的生物标志物多是筛选差异性表达的单个的生物大分子[134].然而,单个蛋白质或者单个基因表达数据的传统统计方法,经常不能识别具有生物意义的生物标志物,导致预测性能低下和有限的临床应用.疾病的发生往往是一群相关联的分子相互作用的结果[135],因此,网络生物标志物作为一种新型生物标志物[136-137]被提出.相比于生物分子标志物,网络生物标志物考虑了生物分子间的关联性,能够更加精确地对病人进行诊断、风险评估等.动态网络生物标志物是在疾病发展过程中,从疾病的不同阶段进行检测和评估,表现为时间依赖性改变的网络生物标志物[138].Chen等人[139-143]提出了一系列的动态网络生物标志物的筛选方法,他们把疾病的发病过程分为3种状态:正常状态(normal state)、发病前状态或临界状态(pre-disease state or critical state)和疾病状态(disease state).正常状态是健康阶段的稳态,疾病尚处在控制中;发病前状态是正常状态的临界点,这个阶段是不稳定的状态,采取合适的治疗,临界状态到正常状态是可逆的;疾病状态就是越过临界点后的另一个稳态,疾病状态到正常状态就不可逆.动态网络生物标志物不仅考虑了生物分子间的关系,还关注了分子网络随时间演化的动态特性,这有助于更全面地精确挖掘生物标识物.基于动态蛋白质网络的生物标志物能够在疾病发展不同的阶段和时间点被监控和估计[138],因此动态网络的生物标志物被认为是有效的检测疾病分水岭相关的基因或蛋白质相互作用的方式之一.Zhang等人[72]基于2组不同发展状态的胶质瘤动态蛋白质网络,发现了与神经胶质瘤预后有关的生物标志物.Li等人[144]识别了与流感、急性肺炎以及二型糖尿病相关的动态蛋白质网络标志物,为揭示复杂疾病的早期诊断以及恶化机制提供了新视角.
5.4 疾病基因预测与疾病研究
大多数疾病可以在基因层面反映出来,现有的一些研究证实了功能相似的基因或者在生物网络中有相互作用关系的基因会导致相同或者相似的疾病[145-149].基于网络的疾病基因识别是发现疾病基因的重要方法,从拓扑结构相似性和功能相似性来分析疾病基因间的关系,对候选基因排序,进而筛选、推断判别出疾病基因[150-152].
相比于静态蛋白质网络,结合蛋白质相互作用数据和基因表达数据构建的动态蛋白质网络能够反映出疾病随时间和外在环境动态变化的过程.在二型的糖尿病研究中,Sun等人[73]通过比较小鼠3个组织的动态网络,发现当发生早期肥胖机能障碍时,肝脏和肌肉组织在整个细胞周期都发生了功能紊乱.Taylor等人[87]研究了人类蛋白质网络的动态结构,并对2组乳腺癌病人进行比较分析,发现人类蛋白质网络的模块化结构的改变将很可能成为乳腺癌预测的一个新指标.Faisal等人[153]研究了年龄特异性的动态网络,结果发现随年龄变化,网络的全局拓扑特性并未有很大的改变,但是局部有改变.与这种变化关联的基因称为年龄相关基因(aging-related genes),这些基因被验证和多种与年龄相关疾病的发生有关系.Yu等人[154]基于动态差异性表达网络研究了糖尿病有关生物通路问题,相比于静态网络,动态网络能够找到更精确的生物通路.
因此,基于动态蛋白质网络识别出疾病基因和疾病通路,有助于疾病治疗药物的开发;同时还能够筛选出更加精确地生物标志物,能够为疾病的诊断和分类提供必要的技术手段.
6 挑战与展望
动态蛋白质网络可以为生物信息学和系统生物学的研究提供一个更综合、更全面的框架.特别是在临床疾病研究和个性化医疗中,为特殊研究目的而构建动态蛋白质网络将变得越来越普及.然而,由于目前能够获得的蛋白质相互作用数据的不完整性,以及基因表达数据中存在的噪声,如何构建更加有效的动态蛋白质网络仍面临诸多挑战.
1) 数据降噪处理
目前常用的动态蛋白质网络构建方法使用的数据主要是PPI数据和基因表达数据,由于诸多因素的影响,这2类数据存在着显著比例的噪声,如何降低蛋白质网络中的假阳性问题?如何分析微阵列基因表达数据中的噪音,提出系统的方法来提取蛋白质表达的动态信息?这些问题都还有待深入研究并解决.
2) 多源数据融合
这里的多源有2种含义:①同种数据类型,不同平台和技术产生的;②不同种数据类型,如多组学数据有PPI数据、基因本体(gene ontology, GO)数据、时间序列RNA-seq数据、时间序列的基因表达数据、亚细胞定位信息、疾病相关数据库等多元信息.目前仍缺乏有效的计算方法能够整合这些数据用于构建更加精确的动态蛋白质网络的研究.如何对不同来源不同类型的数据进行有效的关联分析与整合,构建有效的动态蛋白质网络仍需深入研究.
3) 网络可靠性评估
动态蛋白质网络构建后,一个十分重要的挑战就是如何提出有效的评估指标来判断网络的可靠性,而目前尚未有统一的评价标准.已知的蛋白质网络可靠性评估手段主要分为2个方面:①结合具体的应用(如蛋白质复合物识别等),对结果进行评价.将实验结果和已知数据库中的参考数据相比较,比较不同方法在蛋白质网络上的敏感性和特异性.②对结果做富集性分析,统计显著性水平,说明结果的生物学意义.在实际应用时如何对动态蛋白质相互网络进行量化分析以及如何有效评估动态蛋白质网络的优劣至关重要.
4) 小样本问题
目前动态蛋白质网络构建及后续疾病应用研究都不同程度存在小样本问题.例如,目前能够获得的时间序列的基因表达数据或者RNA-seq数据的时间点一般都比较少,这对于基于蛋白质表达动态性构建方法的准确性有一定的影响.此外,基于多状态下表达及相关性变化的方法往往需要足够的样本才能比较准确地度量2个生物分子间的相关性.在真实数据中,特别涉及到疾病的临床数据时,这样的多样本数据往往是难以获得的,只能获取小样本甚至是单样本数据(如手术前抽血采样等).因此,如何利用能够获取的小样本甚至是单样本数据构建有效的动态蛋白质网络或动态生物分子网络是亟需待解决的重要生物计算问题.
动态蛋白质网络构建是一个循环渐进、不断优化的过程.动态蛋白质网络构建应该以实际应用为目的.目前,动态蛋白质网络已经在蛋白质复合物的识别、蛋白质功能预测、生物标志物识别等方面取得了很好的成果,正逐步应用于特定复杂疾病的分类、早期诊断与后续治疗等.基于复杂网络理论,判断网络是否可控、筛选可靠的药物靶标,将为新的药物设计以及实现精准化医疗提供重要帮助.
[1]Eisenberg D, Marcotte E M, Xenarios I, et al. Protein function in the post-genomic era[J]. Nature, 2000, 405(6788): 823-826
[2]Von Mering C, Krause R, Snel B, et al. Comparative assessment of large-scale data sets of protein-protein interactions[J]. Nature, 2002, 417(6887): 399-403
[3]Albert R, Barabási A L. Statistical mechanics of complex networks[J]. Reviews of Modern Physics, 2002, 74(1): 47-97
[4]Bullmore E, Sporns O. Complex brain networks: Graph theoretical analysis of structural and functional systems[J]. Nature Reviews Neuroscience, 2009, 10(3): 186-198
[5]Pastor-Satorras R, Castellano C, Van Mieghem P, et al. Epidemic processes in complex networks[J]. Reviews of Modern Physics, 2015, 87(3): 925-979
[6]Ocone A, Haghverdi L, Mueller N S, et al. Reconstructing gene regulatory dynamics from high-dimensional single-cell snapshot data[J]. Bioinformatics, 2015, 31(12): i89-i96
[7]Vidalain P O, Boxem M, Ge H, et al. Increasing specificity in high-throughput yeast two-hybrid experiments[J]. Methods, 2004, 32(4): 363-370
[8]Bauer A, Kuster B. Affinity purification-mass spectrometry[J]. European Journal of Biochemistry, 2003, 270(4): 570-578
[9]Hu Bin, Petela N, Kurze A, et al. Biological chromodynamics: A general method for measuring protein occupancy across the genome by calibrating ChIP-seq[J]. Nucleic Acids Research, 2015, 43(20): e132
[10]Stynen B, Tournu H, Tavernier J, et al. Diversity in genetic in vivo methods for protein-protein interaction studies: From the yeast two-hybrid system to the mammalian split-luciferase system[J]. Microbiology and Molecular Biology Reviews, 2012, 76(2): 331-382
[11]Xing S, Wallmeroth N, Berendzen K W, et al. Techniques for the analysis of protein-protein interactions in vivo[J]. Plant Physiology, 2016, 171(2): 727-758
[12]Fukao Y. Protein-protein interactions in plants[J]. Plant and Cell Physiology, 2012, 53(4): 617-625
[13]Zhu H, Qian J. Applications of functional protein microarrays in basic and clinical research[J]. Advances in Genetics, 2012, 79: 123-155
[14]Peng Xiaoqing, Wang Jianxin, Peng Wei, et al. Protein-protein interactions: Detection, reliability assessment and applications[J]. Briefings in Bioinformatics, 2016: bbw066
[15]Li Zhoujun, Chen Yiming, Liu Junwan, et al. A survey of computational method in protein-protein interaction research[J]. Journal of Computer Research and Development, 2008, 45(12): 2129-2137 (in Chinese)
(李舟军, 陈义明, 刘军万, 等. 蛋白质相互作用研究中的计算方法综述[J]. 计算机研究与发展, 2008, 45(12): 2129-2137)
[16]Keskin O, Tuncbag N, Gursoy A. Predicting protein-protein interactions from the molecular to the proteome level[J]. Chemical Reviews, 2016, 116(8): 4884-4909
[17]Papanikolaou N, Pavlopoulos G A, Theodosiou T, et al. Protein-protein interaction predictions using text mining methods[J]. Methods, 2015, 74(2015): 47-53
[18]Badal V D, Kundrotas P J, Vakser I A. Text mining for protein docking[J]. PLoS Computational Biology, 2015, 11(12): e1004630
[19]Salwinski L, Miller C S, Smith A J, et al. The database of interacting proteins: 2004 update[J]. Nucleic Acids Research, 2004, 32(suppl 1): D449-D451
[20]Licata L, Briganti L, Peluso D, et al. MINT, the molecular interaction database: 2012 update[J]. Nucleic Acids Research, 2012, 40(D1): D857-D861
[21]Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life[J]. Nucleic Acids Research, 2015, 43(D1): D447-D452
[22]Kerrien S, Aranda B, Breuza L, et al. The IntAct molecular interaction database in 2012[J]. Nucleic Acids Research, 2012, 40(D1): D841-D846
[23]Pagel P, Kovac S, Oesterheld M, et al. The MIPS mammalian protein-protein interaction database[J]. Bioinformatics, 2005, 21(6): 832-834
[24]Prasad T S K, Goel R, Kandasamy K, et al. Human protein reference database—2009 update[J]. Nucleic Acids Research, 2009, 37(suppl 1): D767-D772
[25]Kotlyar M, Pastrello C, Sheahan N, et al. Integrated interactions database: Tissue-specific view of the human and model organism interactomes[J]. Nucleic Acids Research, 2016, 44(D1): D536-D541
[26]Chatr-Aryamontri A, Breitkreutz B J, Oughtred R, et al. The BioGRID interaction database: 2015 update[J]. Nucleic Acids Research, 2015, 43(D1): D470-D478
[27]Maslov S, Sneppen K. Specificity and stability in topology of protein networks[J]. Science, 2002, 296(5569): 910-913
[28]Jonsson P F, Bates P A. Global topological features of cancer proteins in the human interactome[J]. Bioinformatics, 2006, 22(18): 2291-2297
[29]Bu Dongbo, Zhao Yi, Cai Lun, et al. Topological structure analysis of the protein-protein interaction network in budding yeast[J]. Nucleic Acids Research, 2003, 31(9): 2443-2450
[30]Yook S H, Oltvai Z N, Barabási A L. Functional and topological characterization of protein interaction networks[J]. Proteomics, 2004, 4(4): 928-942
[31]Barabasi A L, Oltvai Z N. Network biology: Understanding the cell’s functional organization[J]. Nature Reviews Genetics, 2004, 5(2): 101-113
[32]Giot L, Bader J S, Brouwer C, et al. A protein interaction map of Drosophila melanogaster[J]. Science, 2003, 302(5651): 1727-1736
[33]Han H W W, Ohn J H H, Moon J, et al. Yin and Yang of disease genes and death genes between reciprocally scale-free biological networks[J]. Nucleic Acids Research, 2013, 41(20): 9209-9217
[34]Watts D J, Strogatz S H. Collective dynamics of ‘small-world’ networks[J]. Nature, 1998, 393(6684): 440-442
[35]Del Sol A, Fujihashi H, O’Meara P. Topology of small-world networks of protein-protein complex structures[J]. Bioinformatics, 2005, 21(8): 1311-1315
[36] Leventhal G E, Hill A L, Nowak M A, et al. Evolution and emergence of infectious diseases in theoretical and real-world networks[J]. Nature Communications, 2015, 6: 6101
[37]Newman M E J, Girvan M. Finding and evaluating community structure in networks[J]. Physical Review E, 2004, 69(2): 026113
[38]Barabási A L, Gulbahce N, Loscalzo J. Network medicine: A network-based approach to human disease[J]. Nature Reviews Genetics, 2011, 12(1): 56-68
[39]Gustafsson M, Nestor C E, Zhang H, et al. Modules, networks and systems medicine for understanding disease and aiding diagnosis[J]. Genome Medicine, 2014, 6(10): 1-11
[40]Sun Huiyan, Liang Yanchun, Chen Liang, et al. An improved sum of edge clustering coefficient method for essential protein identification[J]. Journal of Bionanoscience, 2013, 7(4): 386-390
[41]Jiang Yuexu, Wang Yan, Pang Wei, et al. Essential protein identification based on essential protein-protein interaction prediction by integrated edge weights[J]. Methods, 2015, 83: 51-62
[42]Li Min, Lu Yu, Wang Jianxin, et al. A topology potential-based method for identifying essential proteins from PPI networks[J]. IEEE/ACM Trans on Computational Biology and Bioinformatics, 2015, 12(2): 372-383
[43]Li Min, Lu Yu, Niu Zhibei, et al. United complex centrality for identification of essential proteins from PPI networks[J]. IEEE/ACM Trans on Computational Biology and Bioinformatics, 2017, 14(2): 370-380
[44]Tang Xiwei, Wang Jianxin, Zhong Jiancheng, et al. Predicting essential proteins based on weighted degree centrality[J]. IEEE/ACM Trans on Computational Biology and Bioinformatics, 2014, 11(2): 407-418
[45]Wang Jie, Liang Jiye, Zheng Wenping. A graph clustering method for detecting protein complexes[J]. Journal of Computer Research and Development, 2015, 52(8): 1784-1793 (in Chinese)
(王杰, 梁吉业, 郑文萍. 一种面向蛋白质复合体检测的图聚类方法[J]. 计算机研究与发展, 2015, 52(8): 1784-1793)
[46]Wang Jianxin, Li Min, Deng Youping, et al. Recent advances in clustering methods for protein interaction networks[J]. BMC Genomics, 2010, 11(Suppl 3): S10
[47]Srihari S, Leong H W. A survey of computational methods for protein complex prediction from protein interaction networks[J]. Journal of Bioinformatics and Computational Biology, 2013, 11(02): 1230002
[48]Srihari S, Yong C H, Patil A, et al. Methods for protein complex prediction and their contributions towards understanding the organisation, function and dynamics of complexes[J]. FEBS Letters, 2015, 589(19): 2590-2602
[49]Guo Xingli, Gao Lin, Chen Xin. Models and algorithms for alignment of biological networks[J]. Journal of Software, 2010, 21(9): 2089-2106 (in Chinese)
(郭杏莉, 高琳, 陈新. 生物网络比对的模型与算法[J]. 软件学报, 2010, 21(9): 2089-2106)
[50]Kelley B P, Sharan R, Karp R M, et al. Conserved pathways within bacteria and yeast as revealed by global protein network alignment[J]. Proceedings of the National Academy of Sciences, 2003, 100(20): 11394-11399
[51]Zhang Shihua, Zhang Xiangsun, Chen Luonan. Biomolecular network querying: A promising approach in systems biology[J]. BMC Systems Biology, 2008, 2(1): 5
[52]Xie Jiang, Zhang Wu, Zhang Shihua, et al. A parallel method for querying target subnetwork in a biomolecular molecular network[J]. Int Journal of Numerical Analysis and Modeling, 2012, 9(2): 326-337
[53]Sharan R, Suthram S, Kelley R M, et al. Conserved patterns of protein interaction in multiple species[J]. Proceedings of the National Academy of Sciences, 2005, 102(6): 1974-1979
[54]Clark C, Kalita J. A comparison of algorithms for the pairwise alignment of biological networks[J]. Bioinformatics, 2014, 30(16): 2351-2359
[56]Zhang Xiaochi, Yu Hua, Gong Xiujun. A random walk based iterative weighted algorithm for sub-graph query[J]. Journal of Computer Research and Development, 2015, 52(12): 2824-2833 (in Chinese)
(张小驰, 于华, 宫秀军. 一种基于随机游走的迭代加权子图查询算法[J]. 计算机研究与发展, 2015, 52(12): 2824-2833)
[57]Blum T, Kohlbacher O. MetaRoute: Fast search for relevant metabolic routes for interactive network navigation and visualization[J]. Bioinformatics, 2008, 24(18): 2108-2109
[58]Tian Y, McEachin R C, Santos C, et al. SAGA: A subgraph matching tool for biological graphs[J]. Bioinformatics, 2007, 23(2): 232-239
[59]Wang Jianxin, Peng Xiaoqing, Peng Wei, et al. Dynamic protein interaction network construction and applications[J]. Proteomics, 2014, 14(4/5): 338-352
[60]Tang Xiwei, Wang Jianxin, Liu Binbin, et al. A comparison of the functional modules identified from time course and static PPI network data[J]. BMC Bioinformatics, 2011, 12(1): 339
[61]De Lichtenberg U, Jensen L J, Brunak S, et al. Dynamic complex formation during the yeast cell cycle[J]. Science, 2005, 307(5710): 724-727
[62]Hegde S R, Manimaran P, Mande S C. Dynamic changes in protein functional linkage networks revealed by integration with gene expression data[J]. PLoS Comput Biology, 2008, 4(11): e1000237
[63]Wang Jianxin, Peng Xiaoqing, Li Min, et al. Construction and application of dynamic protein interaction network based on time course gene expression data[J]. Proteomics, 2013, 13(2): 301-312
[64]Shen Xianjun, Yi Li, Jiang Xingpeng, et al. Mining temporal protein complex based on the dynamic PIN weighted with connected affinity and gene co-expression[J]. PLoS ONE, 2016, 11(4): e0153967
[65]Zhang Yijia, Lin Hongfei, Yang Zhihao, et al. Construction of dynamic probabilistic protein interaction networks for protein complex identification[J]. BMC Bioinformatics, 2015, 17(1): 186-186
[66]De Lichtenberg U, Jensen L J, Fausbøll A, et al. Comparison of computational methods for the identification of cell cycle-regulated genes[J]. Bioinformatics, 2005, 21(7): 1164-1171
[67]Tu B P, Kudlicki A, Rowicka M, et al. Logic of the yeast metabolic cycle: Temporal compartmentalization of cellular processes[J]. Science, 2005, 310(5751): 1152-1158
[68]Greenbaum D, Colangelo C, Williams K, et al. Comparing protein abundance and mRNA expression levels on a genomic scale[J]. Genome Biology, 2003, 4(9): 117
[69]Xiao Qianghua, Wang Jianxin, Peng Xiaoqing, et al. Detecting protein complexes from active protein interaction networks constructed with dynamic gene expression profiles[J]. Proteome Science, 2013, 11(S1): S20
[70]Komurov K, White M. Revealing static and dynamic modular architecture of the eukaryotic protein interaction network[J]. Molecular Systems Biology, 2007, 3(1): 110
[71]Xia Kai, Xue Huiling, Dong Dong, et al. Identification of the proliferation/differentiation switch in the cellular network of multicellular organisms[J]. PLoS Computa-tional Biology, 2006, 2(11): e145
[72]Zhang Xiaoyu, Yang Hongbin, Gong Binsheng, et al. Combined gene expression and protein interaction analysis of dynamic modularity in glioma prognosis[J]. Journal of Neuro-Oncology, 2012, 107(2): 281-288
[73]Sun Shaoyan, Liu Zhiping, Zeng Tao, et al. Spatio-temporal analysis of type 2 diabetes mellitus based on differential expression networks[J]. Scientific Reports, 2013, 3(2): 468-473
[74]Shang Xuequn, Wang Yu, Chen Bolin. Identifying essential proteins based on dynamic protein-protein interaction networks and RNA-Seq datasets[J]. Science China Information Sciences, 2016, 59(7): 1-11
[75]Xue Huiling, Xian Bo, Dong Dong, et al. A modular network model of aging[J]. Molecular Systems Biology, 2007, 3(1): 147
[76]Oh S, Song S, Grabowski G, et al. Time series expression analyses using RNA-seq: A statistical approach[J]. BioMed Research International, 2013(3): 203681
[77]Bossi A, Lehner B. Tissue specificity and the human protein interaction network[J]. Molecular Systems Biology, 2009, 5(1): 260
[78]Su A I, Cooke M P, Ching K A, et al. Large-scale analysis of the human and mouse transcriptomes[J]. Proceedings of the National Academy of Sciences, 2002, 99(7): 4465-4470
[79]Su A I, Wiltshire T, Batalov S, et al. A gene atlas of the mouse and human protein-encoding transcriptomes[J]. Proceedings of the National Academy of Sciences, 2004, 101(16): 6062-6067
[80]Boisvert F M, Lam Y W, Lamont D, et al. A quantitative proteomics analysis of subcellular proteome localization and changes induced by DNA damage[J]. Molecular & Cellular Proteomics, 2010, 9(3): 457-470
[81]Zhao Bihai, Wang Jianxin, Li Min, et al. A new method for predicting protein functions from dynamic weighted interactome networks[J]. IEEE Trans on Nanobioscience, 2016, 15(2): 131-139
[82]Hu Sai, Xiong Huijun, Zhao Bihai, et al. Construction of dynamic-weighted protein interactome network and its application[J]. Acta Automatica Sinica, 2015, 41(11): 1893-1900 (in Chinese)
(胡赛, 熊慧军, 赵碧海, 等. 动态加权蛋白质相互作用网络构建及其应用研究[J]. 自动化学报, 2015, 41(11): 1893-1900)
[83]Yang J, Wagner S A, Beli P. Illuminating spatial and temporal organization of protein interaction networks by mass spectrometry-based proteomics[J]. Frontiers in Genetics, 2015, 6: 344
[84]Meng Xiangmao, Li Min, Wang Jianxin, et al. Construction of the spatial and temporal active protein interaction network for identifying protein complexes[C] //Proc of 2016 IEEE Int Conf on Bioinformatics and Biomedicine(BIBM). Piscataway, NJ: IEEE, 2016: 631-636
[85]Tokuriki N, Tawfik D S. Protein dynamism and evolvability[J]. Science, 2009, 324(5924): 203-207
[86]Han J D J, Bertin N, Hao T, et al. Evidence for dynamically organized modularity in the yeast protein-protein interaction network[J]. Nature, 2004, 430(6995): 88-93
[87]Taylor I W, Linding R, Warde-Farley D, et al. Dynamic modularity in protein interaction networks predicts breast cancer outcome[J]. Nature Biotechnology, 2009, 27(2): 199-204
[88]Liu Wei, Xie Hongwei. Construction and analysis of dynamic molecular networks[J]. Progress in Biochemistry and Biophysics, 2014, 41(2): 115-125 (in Chinese)
(刘伟, 谢红卫. 动态分子网络的构建与分析[J]. 生物化学与生物物理进展, 2014, 41(2): 115-125)
[89]Przytycka T M, Singh M, Slonim D K. Toward the dynamic interactome: It’s about time[J]. Briefings in Bioinformatics, 2010, 11(1): 15-29
[90]Hegele A, Kamburov A, Grossmann A, et al. Dynamic protein-protein interaction wiring of the human spliceosome[J]. Molecular Cell, 2012, 45(4): 567-580
[91]Schaefer M H, Lopes T J S, Mah N, et al. Adding protein context to the human protein-protein interaction network to reveal meaningful interactions[J]. PLoS Computational Biology, 2013, 9(1): e1002860
[92]Will T, Helms V. PPIXpress: Construction of condition-specific protein interaction networks based on transcript expression[J]. Bioinformatics, 2016, 32(4): 571-578
[93]Dezsö Z, Nikolsky Y, Sviridov E, et al. A comprehensive functional analysis of tissue specificity of human gene expression[J]. BMC Biology, 2008, 6(1): 49
[94]Fagerberg L, Hallström B M, Oksvold P, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics[J]. Molecular & Cellular Proteomics, 2014, 13(2): 397-406
[95]Lichtenstein I, Charleston M A, Caetano T S, et al. Active subnetwork recovery with a mechanism-dependent scoring function; with application to angiogenesis and organogenesis studies[J]. BMC Bioinformatics, 2013, 14(1): 59
[96]Mitra K, Carvunis A R, Ramesh S K, et al. Integrative approaches for finding modular structure in biological networks[J]. Nature Reviews Genetics, 2013, 14(10): 719-732
[97]Wu G, Stein L. A network module-based method for identifying cancer prognostic signatures[J]. Genome Biology, 2012, 13(12): R112
[98]Zhang W, Ota T, Shridhar V, et al. Network-based survival analysis reveals subnetwork signatures for predicting outcomes of ovarian cancer treatment[J]. PLoS Computational Biology, 2013, 9(3): e1002975
[99]Zhang Xindong, Gao Lin, Liu Zhiping, et al. Identifying module biomarker in type 2 diabetes mellitus by discriminative area of functional activity[J]. BMC Bioinformatics, 2015, 16(1): 92
[100]Bisson N, James D A, Ivosev G, et al. Selected reaction monitoring mass spectrometry reveals the dynamics of signaling through the GRB2 adaptor[J]. Nature Biotechnology, 2011, 29(7): 653-658
[101]Lu Xingyu, Zhao B S, He Chuan. TET family proteins: Oxidation activity, interacting molecules, and functions in diseases[J]. Chemical Reviews, 2015, 115(6): 2225-2239
[102]Hinkelmann F, Brandon M, Guang B, et al. ADAM: Analysis of discrete models of biological systems using computer algebra[J]. BMC Bioinformatics, 2011, 12(1): 295
[103]Killcoyne S, Carter G W, Smith J, et al. Cytoscape: A community-based framework for network modeling[J]. Methods in Molecular Biology, 2009, 563: 219-239
[104]Tang Yu, Li Min, Wang Jianxin, et al. CytoNCA: A cytoscape plugin for centrality analysis and evaluation of protein interaction networks[J]. Biosystems, 2015, 127: 67-72
[105]Bader G D, Hogue C W V. An automated method for finding molecular complexes in large protein interaction networks[J]. BMC Bioinformatics, 2003, 4(1): 2
[106]Wang Jianxin, Zhong Jiancheng, Chen Gang, et al. ClusterViz: A cytoscape APP for cluster analysis of biological network[J]. IEEE/ACM Trans on Computational Biology and Bioinformatics, 2015, 12(4): 815-822
[107]Nepusz T, Yu Haiyuan, Paccanaro A. Detecting overlapping protein complexes in protein-protein interaction networks[J]. Nature Methods, 2012, 9(5): 471-472
[108]Goenawan I H, Bryan K, Lynn D J. DyNet: Visualization and analysis of dynamic molecular interaction networks[J]. Bioinformatics, 2016, 32(17:btw187)
[109]Jeong H, Mason S P, Barabási A L, et al. Lethality and centrality in protein networks[J]. Nature, 2001, 411(6833): 41-44
[110]Wuchty S, Stadler P F. Centers of complex networks[J]. Journal of Theoretical Biology, 2003, 223(1): 45-53
[111]Joy M P, Brock A, Ingber D E, et al. High-betweenness proteins in the yeast protein interaction network[J]. BioMed Research International, 2005, 2005(2): 96-103
[112]Bastian M, Heymann S, Jacomy M. Gephi: An open source software for exploring and manipulating networks[J]. ICWSM, 2009, 8: 361-362
[113]Breitkreutz B J, Stark C, Tyers M. Osprey: A network visualization system[J]. Genome Biology, 2003, 4(3): R22[114]Batagelj V, Mrvar A. Pajek—Analysis and visualization of large networks[G] //LNCS 2265: Graph Drawing Software. Berlin: Springer, 2004: 77-103
[115]Li Min, Tang Yu, Wu Xuehong, et al. C-DEVA: Detection, evaluation, visualization and annotation of clusters from biological networks[J]. Biosystems, 2016, 150: 78-86
[116]Calvano S E, Xiao Wenzhong, Richards D R, et al. A network-based analysis of systemic inflammation in humans[J]. Nature, 2005, 437(7061): 1032-1037
[117]Luo Fei, Liu Juan, Li Jinyan. Discovering conditional co-regulated protein complexes by integrating diverse data sources[J]. BMC Systems Biology, 2010, 4(Suppl 2): S4
[118]Ou-Yang Le, Dai Daoqing, Li Xiaoli, et al. Detecting temporal protein complexes from dynamic protein-protein interaction networks[J]. BMC Bioinformatics, 2014, 15(1): 335
[119]Lei Xiujuan, Wang Fei, Wu Fangxiang, et al. Protein complex identification through Markov clustering with firefly algorithm on dynamic protein-protein interaction networks[J]. Information Sciences, 2016, 329(6): 303-316
[120]Clark W, Radivojac P. Analysis of protein function and its prediction from amino acid sequence[J]. Proteins, 2011, 79(7): 2086-2096
[121]Gaudet P, Livstone M, Lewis S, et al. Phylogenetic-based propagation of functional annotations within the gene ontology consortium[J]. Briefings in Bioinformatics, 2011, 12(5): 449-462
[122]Estrada E, Rodríguez-Velázquez J A. Subgraph centrality in complex networks[J]. Physical Review E, 2005, 71(5): 056103
[123]Li Min, Wang Jianxin, Chen Xiang, et al. A local average connectivity-based method for identifying essential proteins from the network level[J]. Computational Biology and Chemistry, 2011, 35(3): 143-150
[124]Wang Jianxin, Li Min, Wang Huan, et al. Identification of essential proteins based on edge clustering coefficient[J]. IEEE/ACM Trans on Computational Biology and Bioinformatics, 2012, 9(4): 1070-1080
[125]Li Min, Lu Yu, Niu Zhibei, et al. United complex centrality for identification of essential proteins from PPI networks[J]. IEEE/ACM Trans on Computational Biology and Bioinformatics, 2017, 14(2): 370-380
[126]Peng Wei, Wang Jianxin, Wang Weiping, et al. Iteration method for predicting essential proteins based on orthology and protein-protein interaction networks[J]. BMC Systems Biology, 2012, 6(1): 87
[127]Li Gaoshi, Li Min, Wang Jianxin, et al. Predicting essential proteins based on subcellular localization, orthology and PPI networks[J]. BMC Bioinformatics, 2016, 17(Suppl 8): 279
[128]Xiao Qianghua, Wang Jianxin, Peng Xiaoqing, et al. Identifying essential proteins from active PPI networks constructed with dynamic gene expression[J]. BMC Genomics, 2015, 16(Suppl 3): S1
[129]Li Min, Chen Xiaopei, Ni Peng, et al. Identifying essential proteins by purifying protein interaction networks[C] //Proc of Int Symp on Bioinformatics Research and Applications. Berlin: Springer, 2016: 106-116
[130]Greene C S, Krishnan A, Wong A K, et al. Understanding multicellular function and disease with human tissue-specific networks[J]. Nature Genetics, 2015, 47(6): 569-576
[131]Wang Jian, Wu Xiaodan, Chen Luonan, et al. Network biomarkers, interaction networks and dynamical network biomarkers in respiratory diseases[J]. Translational Medicine Research(Electronic Edition), 2014, 4(3): 40-47 (in Chinese)
(王坚, 吴晓丹, 陈罗南, 等. 网络生物标志物, 交互网络和动态网络生物标志物在呼吸系统疾病中的研究进展[J]. 转化医学研究(电子版), 2014, 4(3): 40-47)
[132]Simon R. Development and validation of therapeutically relevant multi-gene biomarker classifiers[J]. Journal of the National Cancer Institute, 2005, 97(12): 866-867
[133]Ludwig J A, Weinstein J N. Biomarkers in cancer staging, prognosis and treatment selection[J]. Nature Reviews Cancer, 2005, 5(11): 845-856
[134]Rhodes D R, Sanda M G, Otte A P, et al. Multiplex biomarker approach for determining risk of prostate-specific antigen-defined recurrence of prostate cancer[J]. Journal of the National Cancer Institute, 2003, 95(9): 661-668
[135]Schadt E E. Molecular networks as sensors and drivers of common human diseases[J]. Nature, 2009, 461(7261): 218-223
[136]Wang Y C, Chen B S. A network-based biomarker approach for molecular investigation and diagnosis of lung cancer[J]. BMC Medical Genomics, 2011, 4(1): 2
[137]Jin Nana, Wu Hao, Miao Zhengqiang, et al. Network-based survival-associated module biomarker and its crosstalk with cell death genes in ovarian cancer[J]. Scientific Reports, 2015, 5: 1566
[138]Wang Xiangdong. Role of clinical bioinformatics in the development of network-based biomarkers[J]. Journal of Clinical Bioinformatics, 2010, 1(1): 28
[139]Chen Luonan, Liu Rui, et al. Detecting early-warning signals for sudden deterioration of complex diseases by dynamical network biomarkers[J]. Scientific Reports, 2012, 2: 342
[140]Liu Rui, Li Meiyi, et al. Identifying critical transitions and their leading biomolecular networks in complex diseases[J]. Scientific Reports, 2012, 2: 813
[141]Liu Rui, Wang Xiangdong, Aihara K, et al. Early diagnosis of complex diseases by molecular biomarkers, network biomarkers, and dynamical network biomarkers[J]. Medicinal Research Reviews, 2014, 34(3): 455-478
[142]Li Meiyi, Zeng Tao, Liu Rui, et al. Detecting tissue-specific early warning signals for complex diseases based on dynamical network biomarkers: Study of type 2 diabetes by cross-tissue analysis[J]. Briefings in Bioinformatics, 2014, 15(2): 229-243
[143]Liu Rui, Yu Xiangtian, Liu Xiaoping, et al. Identifying critical transitions of complex diseases based on a single sample[J]. Bioinformatics, 2014, 30(11): 1579-1586
[144]Li Yuanyuan, Jin Suoqin, Lei Lei, et al. Deciphering deterioration mechanisms of complex diseases based on the construction of dynamic networks and systems analysis[J]. Scientific Reports, 2015, 5: 9283
[145]Liu C C, Tseng Y T, Li Wenyun, et al. DiseaseConnect: A comprehensive Web server for mechanism-based disease-disease connections[J]. Nucleic Acids Research, 2014, 42(W1): W137-W146
[146]Zeng Tao, Zhang Chuanchao, Zhang Wanwei, et al. Deciphering early development of complex diseases by progressive module network[J]. Methods, 2014, 67(3): 334-343
[147]Cho D Y, Kim Y A, Przytycka T M. Network biology approach to complex diseases[J]. PLoS Computational Biology, 2012, 8(12): e1002820
[148]Chakravarti A, Clark A G, Mootha V K. Distilling pathophysiology from complex disease genetics[J]. Cell, 2013, 155(1): 21-26
[149]Hormozdiari F, Penn O, Borenstein E, et al. The discovery of integrated gene networks for autism and related disorders[J]. Genome Research, 2015, 25(1): 142-154
[150]Li Min, Li Qi, Ganegoda G U, et al. Prioritization of orphan disease-causing genes using topological feature and GO similarity between proteins in interaction networks[J]. Science China Life Sciences, 2014, 57(11): 1064-1071
[151]Ganegoda G U,Wang Jianxin, Wu Fangxiang, et al. Prediction of disease genes using tissue-specified gene-gene network[J]. BMC Systems Biology, 2014, 8(Suppl 3): S3
[152]Lan Wei, Wang Jianxin, Li Min, et al. Computational approaches for prioritizing candidate disease genes based on PPI networks[J]. Tsinghua Science and Technology, 2015, 20(5): 500-512

[154]Yu Xiangtian, Zeng Tao, Li Guojun. Integrative enrichment analysis: A new computational method to detect dysregulated pathways in heterogeneous samples[J]. BMC Genomics, 2015, 16(1): 918

Li Min, born in 1978. Professor and PhD supervisor. Member of CCF. She is an awardee of the NSFC Excellent Young Scholars Program in 2016. Her main research interests include bioinformatics, data mining, and deep learning.

Meng Xiangmao, born in 1989. PhD candidate at Central South University. His main research interests include bioin-formatics, complex network analysis, data mining.
The Construction, Analysis, and Applications of Dynamic Protein-Protein Interaction Networks
Li Min and Meng Xiangmao
(SchoolofInformationScienceandEngineering,CentralSouthUniversity,Changsha410083)
The rapid development of proteomics and high-throughput technologies, has produced a large amount of protein-protein interaction (PPI) data, which provides a foundation for further understanding the interactions between proteins and the biomedical mechanism of complex diseases. In an organism, a protein-protein interaction network (PIN) consists of all the proteins and their interactions. Most of the traditional studies on PINs are based on static networks. However, due to the dynamics of protein expressions and the dynamics of PPIs, the real PINs change with time and conditions. Protein function modules related with the occurrence and development of diseases are also bound with this dynamic change. Researchers have shifted their attentions from the static properties to dynamic properties, and proposed a series of methods for the construction of dynamic PINs. This paper is to review the construction, analysis and applications of dynamic PINs. Firstly, the existing dynamic PIN construction methods are discussed in three categories: the methods based on dynamic protein expressions, the methods based on multi-state expression and correlation changes and the methods based on spatial-temporal dynamic changes. The first category embodies the protein dynamic expression varying with time; the second category reflects the changes in the expression-related relationship between proteins under different conditions; while the third category describes the dynamic of proteins and the interactions in time and space. Then, the dynamic analysis of the proteins and the related subnetworks of the dynamic PINs are reviewed. Furthermore,the main applications in the complex diseases of dynamic PINs are discussed in details, such as the identification of protein complexes/functional modules, the detection of biomarkers, and the prediction of disease genes, etc. Finally, the challenges and future research directions of dynamic PINs are discussed.
protein-protein interaction network; dynamic; gene expression; protein complexes; complex diseases
2016-11-25;
2017-03-24
国家自然科学基金优秀青年科学基金项目(61622213);国家自然科学基金项目(61232001,61370024);湖南省研究生科研创新项目(CX2017B063) This work was supported by the National Natural Science Foundation of China for Excellent Young Scientists (61622213), the National Natural Science Foundation of China (61232001, 61370024), and the Hunan Provincial Innovation Foundation for Postgraduate (CX2017B063).
TP399; Q811
