原核表达重组牛凝乳酶的纯化
2017-06-19普燕李轶杰张富春
普燕,李轶杰,张富春
(新疆大学 生命科学与技术学院,新疆生物资源基因工程重点实验室,新疆 乌鲁木齐,830046)
原核表达重组牛凝乳酶的纯化
普燕,李轶杰,张富春*
(新疆大学 生命科学与技术学院,新疆生物资源基因工程重点实验室,新疆 乌鲁木齐,830046)
原核表达获得的重组牛凝乳酶与商品重组牛凝乳酶有相似的酶学特性,具商业潜力,故进一步研究其纯化方法。使用离心与饱和硫酸铵沉淀,成功纯化出有活性的重组牛凝乳酶。蛋白最终回收率为1.82%,总活力比纯化前提高了73.3%,比活力提高了95.3倍。同时,讨论了如何提高包涵体复性率与产物回收率,为今后该酶的工业化生产提供技术参考。
原核表达;重组凝乳酶;纯化;饱和硫酸铵沉淀;回收率;比活力
重组凝乳酶是将动物凝乳酶的cDNA克隆到表达载体中,通过微生物宿主进行表达,经后续分离纯化而获得。美国食品药品管理局(FDA)通过全面审查发表的文献资料得出结论,大肠杆菌K-12表达的重组牛凝乳酶与天然凝乳酶在组分与纯度方面没有差异,且重组凝乳酶制剂中的杂质没有显示出不安全[1]。1990年,该酶成为第一个被FDA批准用于食品的重组酶[2]。现在有3个重组牛凝乳酶符合FDA条例并认为是安全的(GRAS)已被用于干酪生产,它们是EscherichiacoliK-12(大肠杆菌K-12)、kluyveromycesmarxianusvar.lactis(马克斯克鲁维酵母变种)和Aspergillusniger(黑曲霉)表达的重组牛凝乳酶[3]。用这些酶进行Camembert、Cheddar等多种干酪品种的制备,检测干酪出品率、质地、气味、风味和干酪成熟等指标,结果显示这些干酪的重要特性在重组酶与天然牛凝乳酶制备的干酪中没有显著差异,表明重组牛凝乳酶在制备干酪的过程中,其作用方式与天然牛凝乳酶完全相同[1]。如今,估计重组牛凝乳酶占到全世界凝乳酶市场的70%~80%[4]。由此可见,因重组凝乳酶纯度高(含100%凝乳酶)、产量高,以及非特异蛋白水解活性低、凝乳过程的可预测性、符合犹太教规、素食主义者接纳、符合宗教情感和动物权益的保护等特点[1],已经成为小牛皱胃凝乳酶(rennet)的最佳替代品来应对日益增长的干酪市场及伴随的小牛皱胃凝乳酶短缺的问题。
重组凝乳酶在大肠杆菌[5-9]、酵母[3,10]、霉菌[11-12]宿主中均获得成功表达。大肠杆菌能在低廉培养基中高密度快速增殖,且遗传背景清楚,方便克隆表达载体和突变宿主从而被广泛用于外源基因的表达[1]。但也发现,只有转入凝乳酶原cNDA的宿主能最终获得有活性的凝乳酶,直接转入凝乳酶cDNA的宿主表达的蛋白没有活性。凝乳酶原在大肠杆菌中以包涵体形式表达,表达量随着表达系统的改进而不断增加,酶原占总菌蛋白的比例从1%~5%[8]增加到12~19%[7],如今已达66.3%[5]~68%[9],通过包涵体溶解复性和酶原激活步骤,最终获得有活性的成熟的凝乳酶。
凝乳酶原包含365个AA,分子量为40.777 kDa。自然条件下,凝乳酶原在胃的酸性环境下,发生构象改变并自剪切形成有活性的凝乳酶,同时释放N端42个AA(被称为激活片段,activation segment或propeptide),故最后成熟的凝乳酶含有323个AA,分子量为35.6 kDa[1]。体外重组牛凝乳酶原常用的活化方法是:将酶原pH调至pH 2.0,模拟胃的酸性环境,室温放置2 h或更长时间,使酶原发生自剪切产生凝乳酶,再回调pH到pH 6.0左右,进行酶活力测定、酶学特性、纯化等研究。
本实验室原核表达的重组牛凝乳酶的酶学特性与商品化重组凝乳酶(CHY-MAX)没有区别[5],有商业化生产并运用于干酪制作的潜能,故探讨了重组凝乳酶的纯化方法,计算回收率和比活力,为今后该酶在工业上的生产提供技术参考。
1 材料与方法
1.1 材料
1.1.1 菌种与质粒
宿主细胞采用大肠杆菌(Escherichiacoli)菌株BL21 ( DE3),表达载体pET30a-b-proCHY为本实验室构建和保存。
1.1.2 试剂
蛋白 Marker购于北京天根试剂公司和大连宝生物工程有限公司;Bradford法蛋白定量试剂盒购于北京百泰克生物技术有限公司;其他试剂为国产分析纯。
1.2 方法
1.2.1 重组牛凝乳酶原的表达与复性[5]
重组牛凝乳酶原由含有pET30a-b-proCHY的E.coliBL21(DE3)通过IPTG诱导表达。诱导表达1 L菌液,8 000 r/min离心1 min后除去培养基上清,菌体沉淀用20 mL PBS洗涤3次,加18 mL细胞裂解液(50 mmol/L Tris-HCl,2 mmol/L EDTA,pH 8.0),2 mL 10% Triton X-100,加苯甲基磺酰氟(PMSF)至终浓度为1 mmol/L及添加溶菌酶至终浓度1 mg/mL,冰浴超声约30 min,至菌液不再黏稠。12 000 r/min离心20 min,弃去上清,沉淀加20 mL细胞裂解液和2 mL 2%脱氧胆酸,冰浴30 min,期间不断搅拌。12 000 r/min 离心15 min,弃去上清,重复洗涤3次。加20 mL 6 mol/L尿素溶解包涵体,12 000 r/min离心去除不溶杂质,将溶液装入透析袋中,放入约50倍体积的透析液(20 mmol/L Tris-HCl,1 mmol/L EDTA,pH 8.0)中透析,每12 h换1次尿素含量从高向低逐级递减的透析液,最后用不含尿素的透析液透析3次,12 000 r/min离心20 min,上清分装后保存于4 ℃备用。
1.2.2 重组牛凝乳酶原的活化[5]
将透析复性获得的重组凝乳酶原用1 mol/L HCl调至pH 2.0,室温放置2 h,再用1.5 mol/L Tris回调至pH 6.0。此过程中酶原经过酸化自剪切形成有活性的凝乳酶,用于后续纯化及酶活力测定实验。
1.2.3 酶原活化浑浊液的离心处理步骤及不同组分的SDS-PAGE检测
将酶原经酸化/中和处理后得到的浑浊酶液,用12 000 r/min离心10 min,轻缓吸出上清于干净的离心管中,沉淀用无菌蒸馏水重悬(目的是提高凝乳酶的回收率),将重悬溶液离心后取上清,重复2次,将3次上清合并混匀进行后续实验。取适量上清和沉淀样品,进行SDS-PAGE检测,分析上清与沉淀中的组分。
1.2.4 酶原活化浑浊液的自然沉淀处理方法及SDS-PAGE检测
将酸化/中和处理后的浑浊酶液,于4 ℃静置2 h,已经能明显看到分层现象,上层有澄清的趋势,继续放置至24 h,可以得到清亮的上清,轻缓地吸出上清液适量制样(动作要轻,勿使沉淀悬起),继续将上清完全吸出,取适量沉淀制样。上清样和沉淀样进行SDS-PAGE电泳,以检测上清与沉淀中的组分。
1.2.5 饱和硫酸铵沉淀
配制饱和硫酸铵溶液,即在操作温度下,不能再溶解硫酸铵颗粒,过滤去除不溶颗粒备用。按照1.2.3离心方法处理样品,取上清液分装至11个1.5 mL离心管中,每管分装100 μL,分别加入饱和硫酸铵的体积为0、5.3、11.1、17.6、25、33.3、42.9、53.9、66.7、81.8、100 μL,加入饱和硫酸铵时边加边缓慢搅拌,最后吹打混匀,此时得到每管酶液中含饱和硫酸铵比例分别为0、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%。将以上离心管置于4℃ 24 h,取出于4℃、12 000 r/min离心10 min,将上清轻轻移至干净的1.5 mL离心管中,用灭菌蒸馏水补体积至200 μL/管,制样。沉淀加水200μL,混匀后制样。上清样和沉淀样进行SDS-PAGE电泳,以确定凝乳酶沉淀时需达到的饱和硫酸铵浓度。
1.2.6 酶活力的测定[5]
取10 mL 10%的脱脂乳(用0.01 mol/L CaCl2配制),在35℃下保温5 min,加入0.5 mL初步纯化的凝乳酶液,迅速混合均匀,直到在管壁上观察到凝乳颗粒,准确记录从加入酶液到乳液凝固的时间(s),把40 min凝固1 mL 10%的脱脂乳定义为1个索氏单位(Soxhlet unit)。
酶活力计算:
式中:T为乳液凝固时间,s;D为酶液稀释倍数
1.2.7 蛋白定量
按照bradford法蛋白定量试剂盒的说明书操作并计算。
1.2.8 蛋白回收率和酶活提高倍数计算

2 结果与分析
2.1 经酸化/中和处理的重组牛凝乳酶原出现大量沉淀
当对透析离心后的凝乳酶原进行酸化/中和处理时,发现从pH 2.0回调到pH 6.0时出现大量白色沉淀,该沉淀物随放置时间的延长,逐步集中在离心管底部,而上清无色透明,将该液进行离心,结果得到清亮透明上清与沉淀。(见图1)

1-透析后的重组凝乳酶原;2-用1 mol/L HCl将重组凝乳酶原液调至pH2.0;3-用1.5 mol/L Tris-HCl(pH8.8)回调酸化的酶液至pH6.0;4-酸化/中和处理后的酶液于4℃静置2 h;5-浑浊的酶液于12 000 r/min离心10 min图1 重组凝乳酶原经酸化/中和后产生沉淀Fig.1 Recombinant prochymosin was treated by activation/neutralization
2.2 SDS-PAGE检测离心上清与沉淀的组分
将酸化/中和处理的浑浊酶液进行离心(12 000 r/min,10 min),用SDS-PAGE检测离心上清与沉淀,以及检测自然沉淀的酶液(4 ℃静置24 h)上清与沉淀。发现两种方式得到的上清与沉淀组分相同(见图2)。上清中包含凝乳酶和his-propeptide融合肽(见图2中箭头所示),只含有极少量的杂蛋白,沉淀中包含绝大部分细菌杂蛋白以及部分凝乳酶。若将离心处理的沉淀用无菌蒸馏水重悬,再次离心(12 000 r/min,10 min),SDS-PAGE检测上清,发现通过重悬洗涤沉淀,可以从沉淀中溶解出一定量的凝乳酶,这样我们可以通过离心或自然沉淀的方法初步纯化凝乳酶,去除细菌杂蛋白,并且可通过洗涤沉淀来提高凝乳酶的回收率。

1-酶原经酸化/中和处理,浑浊酶液离心后取出上清,沉淀用少量灭菌蒸馏水重悬洗涤,再于12 000 r/min离心10min,取上清检测;2,3-12 000 r/min离心10 min处理浑浊酶液的沉淀与上清;4,5-浑浊酶液静置24 h的沉淀与上清图2 检测酸化/中和处理后浑浊酶液的上清与沉淀组分Fig.2 Detection of the supernatant and pellet of the activation/ neutralization treated recombinant prochymosin
2.3 酶原酸化后回调至不同pH值时上清与沉淀组分检测
重组凝乳酶原经pH2.0酸化2 h,在回调至pH 6.0的过程中,当pH大于3.5时出现白色沉淀,且沉淀量随着pH值的增大(pH 3.5~pH 8.0),先增后减。故将pH2.0酸化酶液回调至不同pH值,经12 000 r/min离心10 min,将上清与沉淀分别制样,进行SDS-PAGE电泳,检测上清与沉淀组分。如图3所示,不同的pH值,上清与沉淀的组分不同。当pH低于3.0时,无沉淀产生;当pH调为pH 3.5时,只有较少的细菌杂蛋白沉淀,凝乳酶和多数细菌杂蛋白均存在上清中;当调到pH4.0时,绝大多数细菌的杂蛋白沉淀而凝乳酶和his-propeptide融合肽仍留在上清中,尤其是pH4.5~pH7.0时,上清中几乎只含凝乳酶和his-propeptide融合肽。但我们也发现,在pH4.0-8.0时,沉淀中始终包含一定量的凝乳酶。其次,回调到不同的pH值,获得的凝乳酶量也不同,在pH5.0附近,上清中的凝乳酶量最低,pH4.5和pH5.5较低,其他pH值比较高。为了兼顾凝乳酶较高的回收率和最佳的酶活力,建议选择pH6.0-6.5作为凝乳酶的初步纯化条件。
2.4 饱和硫酸铵沉淀凝乳酶
酸化/中和处理后的酶溶液经离心初步纯化后,于上清液中加入饱和硫酸铵至不同终浓度,4 ℃放置24 h,12 000 r/min离心10 min,取上清和沉淀制样,进行SDS-PAGE电泳检测,发现当饱和硫酸铵终浓度大于35%时,凝乳酶沉淀而his-propeptide融合肽(约10 kDa)依然留在上清中(图4箭头所示),故选择40%~50%的饱和硫酸铵将凝乳酶与his-propeptide融合肽分离,对凝乳酶进一步纯化。
2.5 重组凝乳酶的纯化步骤
综合上述结果,我们取一定量的凝乳酶原进行酸化/中和处理,12 000 r/min离心10 min,上清吸出保留,沉淀用无菌蒸馏水洗涤并离心,重复2次,合并所有上清,再以饱和硫酸铵终浓度为40%和50%进行沉淀,将各个阶段的样品制样后进行SDS-PAGE电泳,结果见图5。我们可以看到,酶原经酸化/中和处理初期含有很多杂蛋白,而离心后,上清中只包含凝乳酶与his-propeptide融合肽,再经硫酸铵沉淀,凝乳酶能在40%和50%的饱和硫酸铵终浓度下完全沉淀,而his-propeptide融合肽存在于上清中,最终达到了纯化凝乳酶的目的。
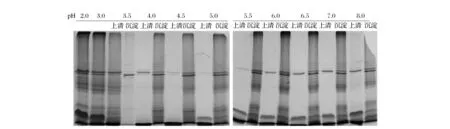
图3 活化酶液回调至不同pH值经离心处理上清与沉淀组分检测Fig.3 Detection of the supernatant and pellet of the activation/neutralization treated recombinant prochymosin in different pH value
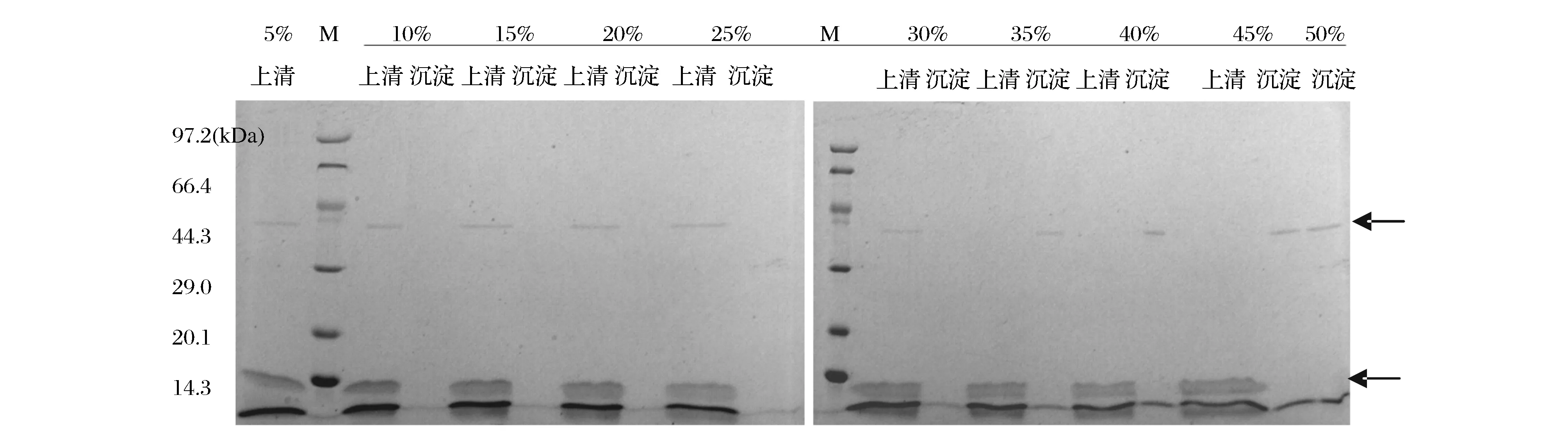
图4 不同饱和硫酸铵终浓度沉淀凝乳酶Fig.4 The different final concentration of saturated ammonium sulfate were used to precipitate chymosin

1-酸化/中和处理的酶原;2-活化酶液离心后上清;3,4-经50%和40%饱和硫酸铵沉淀后离心沉淀物;5,6-经50%和40%饱和硫酸铵沉淀后离心上清图5 重组凝乳酶的纯化步骤Fig.5 The purification of the recombinant prochymosin
2.6 重组凝乳酶在纯化过程中的蛋白回收率与比活力变化情况
用bradford方法测定每一步纯化步骤中蛋白浓度及酶活,结合体积计算出各个时期蛋白回收率,并根据蛋白含量和总酶活计算出凝乳酶的比活力。如表1所示,酶原活化后回调到pH 6.0时的离心上清和硫酸铵沉淀凝乳酶两步相对于原始蛋白含量,蛋白回收率分别为4.93%和1.82%,但总活力却随纯化进行而增加,最后比活力较原始活化酶液提高了27.65和95.33倍。

表1 重组凝乳酶纯化过程中蛋白回收率和比活力变化情况
注:a-酶原经酸化/中和处理得到的浑浊酶液;b-因为浑浊酶液检测蛋白含量不准,故用酶原的测定值来代替;c-将活化酶液离心后,取上清,沉淀经2次洗涤离心,合并3次上清的溶液。
3 讨论
凝乳酶是酶凝干酪制作的关键性酶,随着干酪市场的迅速增长,凝乳酶的用量也不断增加,重组凝乳酶是目前小牛皱胃酶替代品中表现最突出的,因其纯度高,制备干酪的感官指标优于小牛皱胃酶制作的干酪,许多国家都已经自主生产并应用到干酪的制作当中[13-14]。我国目前也非常重视凝乳酶的研发,被列为国家863计划优先发展的项目和国家“七五”、“八五”、“九五”重点攻关项目[14]。
重组凝乳原是最早在大肠杆菌中合成、纯化、鉴定的有活性真核生物蛋白酶之一[8]。从大肠杆菌表达的凝乳酶原占总菌蛋白的5%到我们实验中检测的重组凝乳酶原表达量占总菌蛋白的66.3%[5],这些进步包含了宿主、表达载体等方面的改进。当表达量达到一定程度时,包涵体的复性率和产物的回收率研究就显得迫切了。本研究对原核表达的重组凝乳酶进行纯化,最终产物的回收率为1.82%,对于酶原激活时产生大量沉淀和随着酶的逐步纯化,酶总活力逐步提高的现象,我们进行了以下讨论。
3.1 分析重组凝乳酶原活化过程中产生的沉淀原因
包涵体复性的重组凝乳酶原,在酸化/中和处理前,用高速离心去除杂质,上清透明清亮,用HCl调pH到pH 2.0,溶液一直保持无色透明,但在回调pH到pH 6.0的过程中,出现白色沉淀物(见图1),而且随着pH值的增大,沉淀物的表现也不同,先是沉淀量由少到多,然后是溶液的黏稠度增加,沉淀白色而均匀,最后出现碎絮状沉淀,黏稠度稍有降低,放置一段时间,观察到上清与沉淀的分离。将回调至不同pH值的活化酶液离心,并对上清和沉淀进行SDS-PAGE电泳(见图3),当pH回调到pH 5.0附近时,上清中凝乳酶的含量最低,这也许是因为此时的pH接近重组牛凝乳酶的pI,据报道说牛凝乳酶的pI值为4.6~5.1[1],故重组凝乳酶在pH5.0附近沉淀,在表观上观察到溶液黏稠度增加。当pH增加到6.0以上,凝乳酶溶解度增加,上清中凝乳酶含量也增加。与此同时,我们也观察到沉淀中也有相当含量的凝乳酶,这些凝乳酶可以通过洗涤沉淀的过程回收一部分,原因可能是蛋白总浓度太高,导致一部分凝乳酶随其他杂蛋白一起沉淀下来,可以通过降低活化酶原的浓度来解决。
酶原以包涵体形式在原核细胞中表达,难免包涵体复性透析过程中掺杂一定量的细菌蛋白。一般情况下,绝大多数蛋白不能耐受强酸和强碱的环境,这些细菌蛋白也许因pH2.0的酸化处理而被降解,但此时溶液中存在较多的H+,这些蛋白的表面电荷因带有正电荷而相互排斥,使其保持可溶状态。当回调pH到一定范围时,这些蛋白表面电荷发生变化,从而聚集沉淀。凝乳酶属于天冬氨酸蛋白酶,在酸性条件下稳定,不同种属来源的凝乳酶最适酶活力的pH也均处在酸性环境中,一般在pH5.5~6.5[1],故当杂蛋白都因酸化降解后,凝乳酶依然保持可溶状态并存留于上清中。
我们观察酶原和活化酶液SDS-PAGE电泳图发现[5],若酶原全部自剪切生成凝乳酶,按照分子量大小比例计算,理论上蛋白条带的灰度约为4∶3,但实际观察到酶的条带灰度不及酶原的一半,也就是说部分酶原在激活过程中出现降解。张俊瑞[9]、ESKANDARI[15]等也曾观察到酶原活化产生大量沉淀的现象。猜测可能是酶原复性过程中,仅部分凝乳酶原能正确折叠,这些酶原在活化过程中进行自剪切形成凝乳酶,而错误折叠的酶原不能在酸化时进行自剪切,被酸降解析出。
包涵体的复性率一直是原核表达蛋白的研究重点。若不能正确折叠形成三级结构,那么表达量再高也没有应用价值。目前已经证明凝乳酶原包涵体的复性包括两个阶段,1是要在pH11的缓冲液中完全伸展变性,2是中和到pH8进行复性,而且高pH步骤对酶原复性是必不可少的[16]。WEI等[16]人通过在复性液中添加GroE和10倍的二硫键异构酶(protein disulfide isomerase,PDI)实现在pH8一步法完成复性,其认为GroE和PDI可以有助于凝乳酶原获得天然构象。中国科学院微生物所的杨开宇等人[17],在其发明专利中采用优化的溶解包涵体及再折叠的工艺,可使复性率达到50%左右。唐兵[18]对蛋白质二硫键异构酶(PDI)与GSH/GSSG 促进重组凝乳酶原复性进行了比较,发现二硫键的错误配对是影响凝乳酶原多肽链正确折叠的主要原因。牛凝乳酶中存在3对二硫键[19-20],本实验所采用的酶原复性方法中并没有加入任何能促进二硫键形成的酶或试剂(主要考虑今后工业化生产的成本),故可能也存在二硫键的错配。
3.2 凝乳酶的总活力随着凝乳酶的纯化而增高
表1给我们显示了从酶原活化到凝乳酶最终纯化各步骤中蛋白含量、蛋白浓度、酶活力、总活力和比活力的数据。我们看到凝乳酶总的酶活力和比活力都是增高的。首先,通过离心步骤,总的酶活力上升到了1 889 SU,再经饱和硫酸铵沉淀,总酶活力上升到2 400 SU,比一开始的1 385 SU增高了73.3%。一般酶在纯化过程中,因为存在蛋白的损失和酶的失活,会表现出酶的总活力下降的趋势,但我们纯化过程中却发现凝乳酶的总活力随着纯化的进行而增高,分析可能有以下3种原因。
(1)propeptide的存在干扰了酶的催化作用。凝乳酶在体内是以酶原形式分泌,在胃酸环境中,通过自剪切去除N端42个AA(propeptide)而产生有活性的凝乳酶。将凝乳酶原前体、凝乳酶原和凝乳酶的cDNA克隆到不同的表达载体上,通过不同宿主菌进行表达,发现克隆凝乳酶原cDNA能够成功表达出具有自剪切功能的凝乳酶原,通过酸化/中和处理得到有活性的凝乳酶,而克隆凝乳酶原前体和凝乳酶cDNA往往得不到有活性的凝乳酶,如VALLEJO等[23]克隆了水牛前凝乳酶原、凝乳酶原和凝乳酶序列,只有凝乳酶原克隆表达出了有活性的凝乳酶,这也说明凝乳酶原中的propeptide对凝乳酶的正确折叠有重要作用。YONEZAWA等[21]也证明酶原的N端对整个酶原分子的折叠过程中起决定作用。故目前进行的重组凝乳酶克隆与表达均选用凝乳酶原的cDNA。关于天冬氨酸蛋白酶原的活化过程认为是:propeptide与酶的活性位点以静电作用结合,propeptide的存在阻止了底物进入酶活性中心,故酶原在pH中性环境中不具有催化作用;当pH降低,propeptide与酶活性中心的静电作用被破坏,此时propeptide的构象发生改变,酶原的自剪切位点暴露并与酶活性中心结合,酶对该位点进行切割,propeptide从酶活性中心解离,酶的N端发生构象改变,形成一个反式β折叠位于底物与酶结合的裂隙处[24]。同时,CHRISTENSEN等[25]还发现酶原的活化既可以是分子内的,也可以是分子间的。根据此机制,酶原的活化在体外是通过酸化处理模拟的,通常将pH调到pH 2.0,室温活化2 h,再将pH回调到pH 6.0用于后续的纯化与凝乳活力检测。既然在酶原中,propeptide与酶的活性中心以静电作用方式结合,那么在酶原活化后,如果propeptide和有活性的成熟酶共存于高浓度的溶液中,可能propeptide或多或少会干扰酶与底物的结合,从而降低了酶活力。但Uren等人[26]也同样通过离心和60%饱和硫酸铵沉淀酸化/中和处理后的重组凝乳酶原溶液,其计算的总酶活力是随着纯化步骤的进行逐渐降低的,故是否因为propeptide的存在干扰酶的催化而降低了活力测定值,还需进一步验证。
(2)His标签的存在影响了酶活。一般认为凝乳酶C端区域的负电荷丛与κ-酪蛋白His98-His102正电荷丛之间的静电吸引,对酶与底物的特异结合有重大作用[27]。his标签包含6个串联的his,带正电荷,类似于κ-酪蛋白的His98-His102正电荷丛,可能成为κ-酪蛋白竞争性抑制物从而影响酶活。
(3)纯化去除了溶液中的凝乳酶抑制剂。原核表达系统中是否存在某种物质能够抑制凝乳酶的活性,当其随纯化进行而被去除后,则酶活力提高。NOSEDA等[28]用毕赤酵母GS115表达密码子优化的牛凝乳酶原,发现凝乳酶原自动剪切,产生有活性的凝乳酶上清。将培养基上清先用0.22 μm的滤器过滤,酶活力和总活力以及体积均没有改变,蛋白浓度降低;再进行3 kDa快速超滤,总活力提高到114%,接下来用高效凝胶过滤,总活力回收率为42%,最后再用3 kDa快速超滤,总活力回收率又提高到56%。作者认为纯化过程中出现酶活力提高的情况,推测可能是去除了凝乳酶的假定的抑制剂或蛋白酶之故。
以上3种假设都能解释表1中的离心上清液的酶活力为2 553 SU/mL,而经饱和硫酸铵沉淀后,体积相同的情况下,酶液的酶活力为3 243 SU/mL,提高了27%,但具体原因都还需后续研究验证。
本研究对原核表达获得的重组凝乳酶进行了纯化,使用简单的离心和饱和硫酸铵沉淀的方法获得高纯度、高比活力的凝乳酶。该纯化方法成本低,易操作,对于重组凝乳酶的工业化生产非常有利,然而蛋白的回收率还有提高的空间,如何提高酶原的复性率将是我们下一步研究工作的方向。
[1] KUMAR A,GROVER S,SHARMA J,et al.Chymosin and other milk coagulants: sources and biotechnological interventions[J].Critical Reviews in Biotechnology,2010,30(4): 243-258.
[2] OLEMPSKA-BEER Z S,MERKER R I,DITTO M D,et al.Food-processing enzymes from recombinan microorganisms-a review[J].Regulatory Toxicology and Pharmacology,2006,45(2):144-158.
[4] JACOB M,JAROS D,ROHM H.Recent advances in milk clotting enzymes[J].International Journal of Dairy Technology,2011,64(1):14-33.
[5] 普燕,李轶杰,张富春.原核表达重组牛凝乳酶原及重组牛凝乳酶酶学特性[J].食品与发酵工业,2013, 39(8):13-19.
[7] ZHANG Y,ZHOU Wei,LIU N,et al.Expression of calf prochymosin gene inEscherichiacoli[J].Chinese Journal of Biotechnology,1991,7(3):169-175.
[8] EMTAGE J S,ANGAL S,DOEL M T,et al.Synthesis of calf prochymosin (prorennin) inEscherichiacoli[J].Proc. Nati Acad. Sci.,1983,80:3 671-3 675.
[9] 张俊瑞,马夏吟,张红星,等.牛凝乳酶原基因在大肠杆菌中的高效表达及活性检测[J].中国乳品工业,2012, 40(3): 4-6.
[10] ZHANG L,JIANG Y,ZHANG J,et al.Recombinant expression of bovine chymosin inPichiapastoris[J].Chinese journal of biotechnology,2009, 25(8):1 160-1 165.
[11] MARIANI D D,LORDA G S,BALATTI A P.Rennet production byRhizomucormieheiNRRL 3169[J].Rev Argent Microbiol,2003,35(3):128-132.
[12] TSUCHIYA K,GOMI K,KITAMOTO K,et al.Secretion of calf chymosin from the filamentous fungusAspergillusoryzae[J].Applied Microbiology and Biotechnology,1993,40(2-3):327-332.
[13] 孙海蛟,吕敏,黄艾祥.我国干酪凝乳酶研究及应用现状[J].乳品加工,2008,5:50-52.
[14] 高维东,甘伯中,丁福军,等.微生物凝乳酶的研究进展.中国乳品工业,2009,37(5):34-36.
[15] ESKANDARI M H,HOSSEINI A,ZARASVAND S A,et al.Cloning, Expression, purication and refolding of caprine prochymosin[J].Food Biotechnology,2012,26:143-153.
[16] WEI C,ZHANG Y,YANG K.Chaperone-mediated refolding of recombinant prochymosin[J].Journal of Protein Chemistry,2000,19(6): 449-456.
[17] 杨开宇,刘年娟,张渝英.提高重组牛凝乳酶原的表达与复性的方法: 95105337.X[P]. 1996-02-07.
[18] 唐兵.影响重组凝乳酶原再折叠的主要原因研究[J].武汉大学学报(自然科学版),1997, 43(6):767-770.
[19] ZHANG Y,LI H,WU H, et al.Functional implications of disulfide bond, Cys45-Cys50, in recombinant prochymosin[J]. Biochimica et Biophysica Acta (BBA)-Protein Structure and Molecular Enzymology, 1997,1 343(2):278-286.
[20] CHEN Hong-jie, ZHANG Guo-bao, ZHANG Yu-ying,et al.Functional implications of disulfide bond,Cys206-Cys210,in recombinant prochymosin (Chymosin)[J].Biochemistry,2000,39:12 140-12 148.
[21] YONEZAWA M,SUZUKI J,NISHIYAMA M,et al.Role of the amino-terminal amino acid sequences determining theinvitrorefolding process of prochymosin polypeptide[J].Journal of Biotechnology,1993,28(1):85-97.
[22] CHITPINITYOL S,GOODE D,CRABBE M J C.Studies on the binding of alpha-crystallin to recombinant prochymosins and chymosin[J]. Molecular Vision, 1998, 4: 1. (http://www.molvis.org/molvis/v4/a1/chitpinityol.pdf).
[23] VALLEJO J A,AGEITOS J M,POZA M, et al.Cloning and expression of buffalo active chymosin inPichiapastoris[J].Journal of Agricultural and Food Chemistry,2008,56:10 606-10 610.
[24] RICHTER C,TANAKA T,YADA R Y.Mechanism of activation of the gastric aspartic proteinases: pepsinogen, progastricsin and prochymosin[J].The Biochemical Journal,1998,335:481-490.
[25] CHRISTENSEN K A,PEDERSEN V B,FOLTMANN B.Identification of an enzymatically active intermediate in the activation of porcine pepsinogen[J].FEBS letters,1977,76(2):214-218.
[26] UREN J R,ROBINSON D E,SCANDELLA C J.Recovery and activation process for microbially produced calf prochymosin:4721673[P].1998-1-26.
[27] JENSEN J L,MøLGAARD A,POULSEN J N,et al.Camel and bovine chymosin:the relationship between their structures and cheese-making properties[J].Acta Crystallographica Section D: Biological Crystallography,2013,69(5):901-913.
[28] NOSEDA D G,RECúPEROA M N,BLASCO M, et al.Cloning,expression and optimized production in a bioreactor of bovine chymosin B inPichia(Komagataella)pastorisunder AOX1 promoter[J].Protein Expression and Purification,2013,92(2):235-244.
The purification of the recombinant bovine chymosin expressed in prokaryotic system
PU Yan, LI Yi-jie, ZHANG Fu-chun*
(Xinjiang Key Laboratory of Biological Resources and Genetic Engineering, College of Life Science and Technology,Xinjiang University,Urumqi 830046,China)
The recombinant bovine chymosin expressed in prokaryotic expression system has the similar enzymatic properties to the commercial recombinant bovine chymosin, so it has the potential to be commercialized. Therefore the purification of the enzyme was studied. The recombinant bovine chymosin was purified successfully by centrifugation and saturated ammonium sulfate precipitation. The ultimate recovery is 1.82%. The total activity increased by 73.3% and the specific activity increased 95.3-fold. How to improve the renaturation of inclusion body and recovery also were discussed, which provided technical reference for industrial production of the enzyme in the future.
prokaryotic expression; recombinant chymosin; purification;saturated ammonium sulfate precipitation; recovery; specific activity
10.13995/j.cnki.11-1802/ts.201704010
博士,讲师(张富春教授为通讯作者,E-mail:zfcxju@gmail.com)。
新疆维吾尔自治区自然科学基金青年基金(2013211B10)
2016-10-16,改回日期:2016-11-27
