绿原酸溶液、微乳和凝胶的透皮特性与皮肤刺激性研究
2017-06-15侯成贤杨燕飞
陈 莉,侯成贤,杨燕飞
(遵义医学院 药学院药剂学教研室,贵州 遵义 563099)
基础医学研究
绿原酸溶液、微乳和凝胶的透皮特性与皮肤刺激性研究
陈 莉,侯成贤,杨燕飞
(遵义医学院 药学院药剂学教研室,贵州 遵义 563099)
目的 对绿原酸溶液、微乳和凝胶的体外透皮特性和皮肤刺激性进行对比研究,为绿原酸外用制剂的开发提供实验依据。方法 以雄性大鼠背部皮肤为渗透屏障,采用Franz扩散池比较不同制剂中绿原酸的透皮特性,并采用大鼠自身对照考察绿原酸不同制剂对大鼠皮肤的刺激性。结果 绿原酸3种制剂的累积渗透量Qn与时间t有较好线性关系。微乳及凝胶组24 h的累积渗透量Q24h分别为溶液组的7.71倍和7.49倍,其透皮速率常数J分别为7.98倍和7.50倍,且累积透过百分率分别为2.63倍和2.16倍。绿原酸凝胶组的体外透皮释放过程符合Higuchi模型。绿原酸制剂对大鼠皮肤均无刺激性。结论 绿原酸微乳及凝胶能够促进绿原酸经皮渗透,有望开发成为新的绿原酸外用制剂。
绿原酸;微乳;凝胶;体外经皮渗透;皮肤刺激性
绿原酸是植物体在有氧呼吸过程中由咖啡酸与奎尼酸生成的缩酚酸,具有较广泛的抗菌、抗氧化、抗衰老、抗肿瘤、降压、补肾、利尿、利胆等作用[1],口服生物利用度低[2-3]。因其分子量小、油水分配系数适宜[4]、毒性低,可将其制成经皮给药制剂,避免肝脏首过效应及被肠道菌群破坏[5],延长给药时间,提高局部或全身治疗的疗效,便于使用。
微乳是经皮给药制剂有前景的载体,对药物具有增溶、促渗及增加皮肤滞留量的作用[6],可通过改变角质层的脂质、促进角蛋白水化及改变角蛋白结构从而降低其屏障功能[7]等促渗机制,提高给药的有效性与安全性。此外,水凝胶剂也是近年来发展较快的一种新型经皮给药剂型,具有分散性好、黏附力强、稳定性好、药效持久、应用方便、透皮吸收快、可避开首过效应和给药部位浓度高等优点,与外用溶液剂相比,还可延缓药物释放,延长药物在皮肤上的保留时间[8]。有关绿原酸溶液、微乳和凝胶的透皮给药目前鲜见报道,因此本文重点考察三者的透皮性质以及对大鼠皮肤的刺激性,为研究绿原酸新制剂及新的给药途径奠定基础。
1 材料与方法
1.1 试药 绿原酸对照品,中国药品生物制品检定所,批号:110753-200413;绿原酸原料药(金银花提取物),96%,南京朗泽医药科技有限公司;油酸乙酯,阿拉丁试剂公司;吐温-80,重庆川江化学试剂厂;丙三醇,成都市科龙化工试剂厂;卡波姆940,郑州优然食品化工有限公司;甲醇,色谱纯,上海跃胜贸易有限公司;乙腈,色谱纯,上海跃胜贸易有限公司;磷酸,分析纯,成都市科龙化工试剂厂。
1.2 仪器 Agilent 1 260高效液相色谱仪,美国安捷伦有限公司;TGL-16 C高速台式离心机,上海安亭科学仪器厂;XW-80 A涡旋混合器,海门市其林贝尔仪器制造有限公司;ME 204 E万分之一分析天平,梅特勒-托利多仪器(上海)有限公司;TK-24 BL透皮扩散试验仪(接收池体积为8 mL,有效透皮面积为3.14 cm2),上海锴凯科技贸易有限公司;DN-36 A氮吹仪,无锡沃信仪器有限公司。
1.3 实验动物 健康雄性SD大鼠,SPF级,体重(200±20) g,购自重庆第三军医大学医学实验中心,许可证号:SCXK(渝)2012-0005,适应性饲养1周。动物房温度为(23±2) ℃,相对湿度为50%±10%,每日照明12 h,大鼠给药后采用单笼饲养,自由饮食和饮水。1.4 色谱条件 DIKMA Plastisill C18色谱柱(4.6 mm×250 mm,5 μm ),并配以DIKMA C18(10 mm×4.6 mm)保护柱;流动相:乙腈-0.4%磷酸(15∶85,v/v);检测波长:327 nm;流速:1.0 mL/min;柱温:25 ℃;进样量:20 μL。
1.5 方法学考察
1.5.1 方法专属性 取空白接收液、空白接收液+对照品及透皮试验样品,经0.22 μm微孔滤膜过滤,取续滤液20 μL进样分析。
1.5.2 标准曲线的制备 精密称取绿原酸对照品11.67 mg,置于50 mL棕色容量瓶中,用50%甲醇溶液溶解并定容至刻度,摇匀,得浓度为233.33 μg/mL的对照品储备液。再用50%甲醇溶液依次稀释成浓度为23.33、11.67、5.83、2.92、1.46、0.73、0.36、0.18、0.09 μg/mL的标准系列溶液。经0.22 μm微孔滤膜过滤后进样。以绿原酸的峰面积为纵坐标(Y),标准系列浓度(μg/mL)为横坐标(X),绘制标准曲线,得出标准曲线方程和线性。
1.5.3 精密度试验 分别制备绿原酸低(0.36 μg/mL)、中(2.92 μg/mL)、高(11.67 μg/mL)3个浓度的对照样品,过滤后重复进样6次并连续测3 d。将峰面积带入标准曲线方程计算各样品的浓度,求得日内和日间精密度(RSD%)。
1.5.4 加样回收率试验 按1.5.3项下方法制备绿原酸低、中、高3个浓度的对照样品各3份,分别加入等量另一已知浓度的供试品,混匀过滤后进样,测得相应峰面积,带入标准曲线求得混合样品浓度,计算加样回收率。
1.5.5 稳定性试验 分别于0、1、3、6、9、12、18、24、30 h测定同一透皮试验样品的峰面积,考察待测样品在室温放置30 h的稳定性(RSD%)。
1.6 透皮制剂的制备
1.6.1 绿原酸溶液 精密称取绿原酸原料药0.054 5 g加10 mL纯水搅拌溶解,即得绿原酸浓度为5.420 5 mg/g(w/w)的无色澄明溶液。
1.6.2 绿原酸微乳 按照文献比例(1∶3∶6,w/w/w)[9],精密称取油酸乙酯0.10 g、吐温-80 0.30 g和甘油0.60 g,混匀后加入绿原酸原料药0.100 5 g于37 ℃水浴中搅拌溶解完全,在搅拌下缓慢加入19 mL纯水使之完全乳化,即得绿原酸浓度为4.999 9 mg/g(w/w)的无色绿原酸微乳。1.6.3 绿原酸凝胶剂 精密称取卡波姆940 0.16 g,加甘油2.00 g润湿后再加入8 mL纯水混匀待其完全溶胀。另精密称取绿原酸原料药0.109 1 g加10 mL纯水搅拌溶解后与完全溶胀的卡波姆940混合均匀,加适量三乙醇胺调pH至5.5[4],即得绿原酸浓度为5.382 6 mg/g(w/w)的无色透明黏度适宜的绿原酸凝胶剂。
1.7 绿原酸溶液、微乳和凝胶的体外透皮考察
1.7.1 大鼠离体皮肤的制备 取健康雄性大鼠脱毛膏脱去背部毛发,饲养24 h后颈部脱臼处死,用手术剪小心剥离背部皮肤,铺于玻璃板上仔细剔除皮下脂肪层和结缔组织,用生理盐水反复冲洗干净,并于-20 ℃中保存备用。实验前自然解冻,再用生理盐水浸泡15 min,取完整皮肤进行实验。
1.7.2 透皮扩散实验 采用Franz扩散池法。将大鼠背部皮肤固定于透皮扩散仪的供给池和接收池之间,角质层朝向供给池。在供给池中分别加入绿原酸溶液、微乳及凝胶各1.00 g,接收池中加入生理盐水8 mL作接收液。磁搅拌转速为300 rpm,温度为32 ℃,分别于0.5、1、1.5、3、5、7.5、10、12和24 h从接收池取出接收液1 mL,并立即补充等体积同温度的接收液。样品经0.22 μm微孔滤膜过滤后用HPLC法测定绿原酸的含量。按下式计算各个时间点的单位面积累积透过量(Qn,μg/cm2)[10]。
Qn=(CnVn+∑CiVi)/A
式中,Cn为第n个取样点的浓度(μg/mL);Ci为第i个取样点的浓度(μg/mL);Vn为接收池体积(8 mL);Vi为第i个取样点取样体积(1 mL);A为有效透皮面积(3.14 cm2)。以Qn对时间t作图,并对曲线中的直线部分线性回归,求出直线斜率,即稳态渗透速率J[μg/(cm2·h)]。
1.8 大鼠皮肤刺激性试验 健康雄性SD大鼠36只,按体重随机均分为两组:完整皮肤组和破损皮肤组。每组再按受试物不同随机分为3组:溶液组、微乳组和凝胶组。在给药前去除大鼠背脊两侧毛发,面积约5 cm×10 cm,破损皮肤组除毛后需用砂纸磨擦,去毛部位以渗血为度。以大鼠自身左侧皮肤为对照,于右侧去毛区皮肤均匀涂抹制剂1.00 g。每天给药1次,连续7 d,观察受试期间大鼠行为活动、精神状况及饮食情况。于末次给药4 h后用温水洗去残留受试物,观察第1、24、48、72小时大鼠涂抹部位有无出现红斑、水肿等情况,并按表1中的评价方法进行刺激性评分,计算出平均值后根据以下评价标准进行评价。无刺激性:0~0.49;轻度刺激性:0.50~2.99;中度刺激性:3.00~5.99;强刺激性:6.00~8.00[11]。
表1 皮肤刺激性反应评分标准

刺激反应的表征(红斑)分值刺激反应的表征(水肿)分值无红斑0无水肿0轻微红斑(勉强可见)1轻微水肿(勉强可见)1中度红斑2中度水肿(涂药区域隆起轮廓明显)2严重红斑3重度水肿(涂药区域隆起约1mm,轮廓清楚)3紫红色红斑并有焦痂形成4严重水肿(涂药区域隆起约1mm,并范围扩大)4

2 结果
2.1 方法学考察结果 结果见图1和表2。绿原酸的保留时间为8.5 min左右,生理盐水及透皮试验中的其他杂质对绿原酸的测定无干扰。绿原酸的标准曲线方程为:Y=28.72X+0.615,R2=0.999 9,表明绿原酸为0.09~23.33 μg/mL时,峰面积与浓度线性关系良好。精密度、回收率结果符合绿原酸含量测定要求。透皮试验样品在室温放置30 h的RSD%为5.47,表明样品在30 h内含量稳定。综上所述,本文所建立的色谱方法适于透皮试验样品中绿原酸的含量测定。

A:空白接收液;B:空白接收液+绿原酸对照品;C:绿原酸供试品;1:绿原酸。图1 绿原酸HPLC图
表2 精密度和方法回收率试验结果

浓度(μg/mL)精密度(RSD%)日内(n=6)日间(n=3)方法回收率(%)x±sRSD0.365.436.8790.35±6.066.712.933.205.0393.33±5.065.4211.674.435.2492.03±3.754.07
2.2 透皮试验结果 将绿原酸溶液、微乳及凝胶各时间点的累积渗透量Qn分别对时间t进行线性回归,得到渗透动力学方程,所得斜率为透皮速率常数J[μg/(cm2·h)](见表3)。绿原酸溶液、微乳及凝胶的累积渗透量Qn与渗透的时间t有较好的线性关系,采用t检验进行统计分析,与绿原酸溶液组比较,微乳及凝胶组的透皮渗透速率J与累积渗透量Q24h有明显差异(P<0.05),其中透皮速率常数J分别是溶液组的7.98倍和7.50倍,而累积渗透量Q24 h分别是溶液组的7.71倍和7.49倍。
对绿原酸凝胶剂的透皮数据分别采用零级模型、一级模型和Higuchi模型拟合[12]。从拟合结果(见表4)可看出,Huguchi方程的R2最大,其次是零级方程,最小的是一级方程,因此,绿原酸凝胶的体外透皮释放过程更符合Higuchi模型,故推测绿原酸在凝胶中具有缓释特征。绿原酸3种制剂的累积渗透曲线见图2。5 h以前,三者累积透过率均较低(P>0.05),但5 h之后绿原酸微乳与凝胶的透过率明显高于绿原酸溶液(P<0.05)。3种制剂中绿原酸的24 h透过率分别为1.27%、3.34%和2.74%,其中微乳组和凝胶组的透过率分别是溶液组的2.63倍和2.16倍。
表3 绿原酸不同制剂的渗透动力学方程(n=6)

组别渗透动力学方程(Q-t)R2J[μg/(cm2·h)]Q24h(μg/cm2)绿原酸溶液Q=6.016t+9.96080.91146.016134.185绿原酸微乳Q=48.006t-113.150.999548.006*1034.722*绿原酸凝胶Q=45.139t-59.0440.991745.139*1005.510*
*:vs绿原酸溶液,P<0.05。
表4 绿原酸凝胶剂体外累积释放模型拟合

名称方程R2零级方程(Q-t)Q=44.544t-51.1120.9917一级方程(lnQ-t)lnQ=0.0707t+5.26170.9439Higuchi方程(Q-t1/2)Q=331.56t1/2-625.070.9995
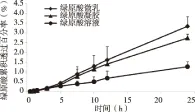
图2 绿原酸在不同制剂中的累积渗透曲线
2.3 大鼠皮肤刺激性试验 根据表1中的评价方法考察大鼠涂抹部位的红斑和水肿情况,结果显示大鼠给予绿原酸3种制剂后,行为活动、精神状况、饮食情况均正常,去除受试物后无论是完整皮肤还是破损皮肤均未见红斑、水肿等皮肤刺激性表征(各时间点评分结果为0),表明绿原酸3种制剂对皮肤均无刺激性。
3 讨论
本文透皮试验采用了大鼠背部皮肤,较常用的小鼠或大鼠的腹部皮肤更厚,然而透皮试验结果显示,3种制剂的绿原酸都能透过该皮肤,并随着时间的延长呈线性增加。三者的透皮能力依次为:绿原酸微乳>绿原酸凝胶>绿原酸溶液剂。
绿原酸溶液剂的经皮透过性可能与其理化性质[13]有密切关系:①绿原酸相对分子质量为354.31(小于500),较易穿透皮肤屏障。②表皮的pH值为4.2~5.6,在此生理条件下绿原酸能够以较稳定的分子形式存在,较离子型有更好的透过性。③研究表明,lgP<1或者lgP>5的化合物吸收都较差,而绿原酸25 ℃的表观油水分配系数lgP为1.42[4](大于1且小于5),也为绿原酸的经皮渗透提供了有利条件。与溶液剂相比,绿原酸微乳的渗透速率和渗透量明显增加,表明微乳有利于药物经皮吸收。此结果可能与微乳的特性有关:①绿原酸微乳粒径小(约20 nm[9]),有利于降低表面张力,湿润皮肤,保证药物的经皮渗透[14]。②改善药物在皮肤角质层的分配系数,渗透系数增大,相对于绿原酸水溶液更容易通过角质层并滞留在皮肤内形成贮库[15]。③微乳处方中的表面活性剂(吐温-80)可以改变生物膜的通透性,增加细胞间隙,促进药物渗透。④提高药物稳定性,增加药物的溶解度,增大药物在皮肤两侧的浓度梯度,起到促渗剂的作用[16]。绿原酸微乳具有很强的透皮能力,有望进一步开发成为新的绿原酸外用制剂,为绿原酸新剂型及新制剂进一步开发研究提供实验依据。
此外,绿原酸凝胶剂以卡波姆940为凝胶基质并用三乙醇胺调节pH值至5.5,在此条件下凝胶黏度适宜,绿原酸性质稳定[4],能够以分子形式均匀分散于基质的网状结构中,为绿原酸的经皮渗透提供了有利条件。与溶液剂比较,凝胶剂还具有较强的皮肤黏附和润湿能力,从而使得绿原酸凝胶的透皮能力优于溶液剂。然而凝胶剂的促渗作用略低于微乳,其具体的促渗机制还有待进一步研究。考虑到微乳经皮给药存在黏附性差、滞留时间较短等问题,拟将其进一步制备成微乳凝胶,并考察微乳与凝胶联合应用对药物透皮吸收是否存在协同促进作用。
[1] 霍晓芳,唐彦萍,张庆军,等.绿原酸对脂多糖诱导的巨噬细胞的影响[J].遵义医学院学报,2003,26(6):507-510.
[2] de Oliveira D M,Sampaio G R,Pinto C B,et al.Bioavailability of chlorogenic acids in rats after acute ingestion of maté tea (Ilex paraguariensis) or 5-caffeoylquinic acid[J].Eur J Nutr,2016,doi:10.1007/s00394-016-1290-1.
[3] Jung J W,Kim J M,Jeong J S,et al.Pharmacokinetics of chlorogenic acid and corydaline in DA-9701,a new botanical gastroprokinetic agent,in rats[J].Xenobiotica,2014,44(7):635-643.
[4] 毕肖林,李瑶瑶,杜秋,等.绿原酸表观油水分配系数测定及在体肠吸收动力学研究[J].南京中医药大学学报,2013,29(6):572-575.
[5] 何心,赵铁敏,郡修德,等.绿原酸的药代动力学研究[J].中成药,1999,21(4):161-162.
[6] 范永春,戴薇,李步阳.蟾蜍毒素微乳的制备及体外透皮吸收考察[J].中国实验方剂学杂志,2013,19(21):49-53.
[7] 梅芬,曾维东,万涛,等.醋酸地塞米松微乳的制备及对皮肤渗透性的影响[J].药学学报,2016,51(6):979-984.
[8] 钟华,尹蓉莉,谢秀琼,等.莪术微乳凝胶的制备工艺设计与优化[J].成都中医药大学学报,2012,35(3):72-75.
[9] 陈莉,周鑫,申登峰.绿原酸自微乳给药系统的处方设计与体外评价[J].遵义医学院学报,2015,38(4):387-391.
[10] 蒋楠,孙雯,李晔,等.葛根素微乳的制备及体外透皮特性研究[J].西北药学杂志,2013,28(2):180-183.
[11] 王敏,林茂,邓英杰,等.盐酸丁卡因脂质体凝胶皮肤刺激性实验及药效[J].遵义医学院学报,2015,38(6):599-603.
[12] 管咏梅,赵益,陈丽华,等.雷公藤微乳凝胶释药性能研究[J].中国实验方剂学杂志,2010,16(17):1-3.
[13] 方亮.药剂学[M].北京:人民卫生出版社,2016:240.
[14] 王正,慕宏杰,张峰溥,等.罗替戈汀微乳凝胶的制备与体内外评价[J].中国新药杂志,2015,24(8):930-941.
[15] Xiong X Y,Li J.Studies on the microemulsion-based hydrogel formulation of ketoprofen and the transdermal mechanism[J].Journal of China Pharmaceutical University,2012,43(6):514-518.
[16] 魏文珍,陈晓兰,吴玉梅,等.雪上一枝蒿微乳体外透皮及影响因素研究[J].时珍国医国药,2015,26(9):2132-2134.
[收稿2016-12-26;修回2017-02-07]
(编辑:王静)
Study on in vitro transdermal permeability and skin irritation of chlorogenic acid in solution, microemulsion and carbomer hydrogel
ChenLi,HouChengxian,YangYanfei
(Department of Pharmaceutics, School of Pharmacy, Zunyi Medical University, Zunyi Guizhou 563099, China)
Objective To investigate the differences in transdermal permeability and skin irritation of chlorogenic acid in three different formulations based on solution, microemulsion and carbomer hydrogel. Methods Franz diffusion cells were used for the transdermal permeation study using male rats dorsal skin. Irritation effects on rats intact or damaged skin were studied by self control. Results The accumulative amount of the permeated chlorogenic acid (Qn) increased linearly with time (t). Within 24 hours, theQ24hof chlorogenic acid in microemulsion and gel groups were 7.71 times and 7.49 times than that in solution group, respectively. The permeation rate (J) was 7.98 times and 7.50 times, meanwhile the accumulative permeation percent ratio was 2.63 times and 2.16 times. Theinvitrotransdermal delivery ability of chlorogenic acid in gel group conformed to Higuchi equation. The skin irritation test indicated that chlorogenic acid formulations did not cause irritation on the intact or damaged skin.Conclusion Chlorogenic acid formulations have no stimulation to rat skin. Compared with the solution group, microemulsion and gel groups can promote transdermal permeability, suggesting that microemulsion and gel may be promising new transdermal formulations of chlorogenic acid.
chlorogenic acid; microemulsion; gel; transdermal permeation; skin irritation
贵州省联合基金资助项目(NO:黔科合J字LKZ[2013]26)。
R943
A
1000-2715(2017)02-0119-05
