OC-STAMP基因过表达载体及稳转细胞系的构建
2017-06-15袁慧敏何茳萍张光亚张丹丹陈凤玲
袁慧敏,何茳萍,张光亚,张丹丹,陈凤玲
(1.蚌埠医学院 临床学院,安徽 蚌埠 233030; 2.上海交通大学医学院附属第九人民医院 内分泌科,上海 201900)
基础医学研究
OC-STAMP基因过表达载体及稳转细胞系的构建
袁慧敏1,何茳萍2,张光亚2,张丹丹2,陈凤玲2
(1.蚌埠医学院 临床学院,安徽 蚌埠 233030; 2.上海交通大学医学院附属第九人民医院 内分泌科,上海 201900)
目的 构建破骨细胞多次跨膜蛋白(OC-STAMP)基因过表达质粒,体外转染RAW264.7细胞构建稳转细胞系,为进一步研究OC-STAMP的功能提供工具。方法 根据NCBI数据库中OC-STAMP基因信息,设计引物,采用PCR扩增其基因片段; 运用基因重组方法双酶切目的基因,并将其克隆至含有绿色荧光蛋白(GFP)的pCDH-CMV慢病毒载体,经酶切测序对重组质粒进行鉴定;转染成功构建的质粒至293T细胞中Western blotting验证蛋白过表达情况;采用psPAX2和pMD2.G两种包装质粒包装pCDH-CMV-oc-stamp重组质粒获得病毒液,病毒感染RAW264.7细胞获得OC-STAMP过表达稳转细胞系,Q-PCR和Western blotting验证蛋白过表达情况。结果 酶切和测序鉴定表明成功构建pCDH-CMV-oc-stamp的基因重组质粒; 将成功构建的pCDH-CMV-oc-stamp质粒转染至293T,可观察到大量绿色荧光表达; 将成功包装获得的病毒液感染RAW264.7细胞后q-PCR及Western blotting结果证实OC-STAMP成功过表达。结论 成功构建pCDH-CMV-oc-stamp重组质粒及OC-STAMP过表达的稳转细胞系。
破骨细胞多次跨膜蛋白;质粒构建;病毒转染;RAW264.7细胞
破骨细胞跨膜蛋白(Osteoclast stimulatory transmembrane protein,OC-STAMP) 是Yang等[1]于2008年在经核因子B受体活化因子配体( receptor activator of nuclear factor-Bligand,RANKL) 诱导后的小鼠巨噬细胞株RAW 264.7细胞及原代骨髓巨噬细胞中发现的一个多次跨膜蛋白。小鼠的OC-STAMP基因位于2号染色体上,与人的同源性和相似性分别为74%和86%。该基因结构上高度保守,蛋白表达广泛,且具有组织特异性,已经证实OC-STAMP在肝脏、心脏及脑等组织中高表达,这提示OC-STAMP可能具有重要的生理功能[2]。OC-STAMP作为近年来新发现的细胞融合相关蛋白分子,主要调控破骨细胞融合和异物巨噬细胞融合[3-16],但关于OC-STAMP的功能研究仍不明确。在我们的前期工作中,利用Affymetrix mouse 430 2.0基因表达谱芯片杂交技术观察到小鼠OC-STAMP基因在糖尿病模型小鼠db/db鼠的腹腔静置巨噬细胞中表达明显下调,其表达量是同窝野生型对照组小鼠的1/13,并在小鼠原代腹腔静置巨噬细胞mRNA水平也验证了以上结果。同时我们成功制备了OC-STAMP的多克隆抗体[17]。考虑到OC-STAMP基因是一个功能尚不清楚的基因,且在小鼠糖尿病代谢状态中有明显变化,因此我们设想糖尿病状态的一些代谢因素(如高血糖、高血脂、炎症因子等)可降低OC-STAMP的表达水平,OC-STAMP可能参与相关代谢环节。为进一步研究OC-STAMP的蛋白功能及其对糖尿病的代谢状态的可能影响,我们拟构建OC-STAMP过表达载体及稳转细胞系,为后期的功能机制研究奠定基础,提供研究工具。
1 材料与方法
1.1 试剂与仪器
1.1.1 主要试剂 大肠埃希菌菌株DH 5 购于康为世纪公司; 限制性内切酶Nhe I/Not I、T 4连接酶、Q 5高保真聚合酶均购自NEB公司; 所用引物均订购于上海生工有限公司;5 000 bp DNA maker购于上海业立生物技术有限公司; 质粒大抽提试剂盒购自上海天根生物科技公司;小质粒抽提试剂盒购于TaKaRa公司;转染试剂Lipofectamine 3 000购自Invitrogen公司;Polybrene及Puromycin购自Sigma公司;小鼠单核细胞株RAW 264.7细胞购自ATCC细胞库;293 T细胞受赠于上海交通大学医学院附属第九人民医院烧伤科实验室。
1.1.2 主要仪器 超净工作台(海尔A 2生物),倒置荧光相差显微镜(Olympus,日本),低温离心机(Eppendorf,德国),PCR 基因扩增仪(ABI 公司,美国),Bio-RAD电泳仪和琼脂糖水平电泳槽(BIO-RAD),Fusion FX 7图像扫描分析仪器(Viber,法国),隔水恒温振荡箱(培英实验设备有限公司),隔水式恒温培养箱(上海精宏实验设备有限公司),超滤管(Millipore,美国),10 cm2/6 cm2/3 cm2细胞培养皿、15 mL/5 mL离心管、6孔板、 96孔板(Corning,美国),1 000 μL/100 μL/10 μL/2.5 μL移液枪(Eppendorf,德国)。
1.2 方法
1.2.1 引物设计与合成 根据NCBI数据库中的鼠OC-STAMP基因编码区( CDS区)序列,经PrimerPremier 5分析,并依据质粒载体pCDH-CMV的多克隆位点设计相应引物; 在引物前分别添加Nhe I/Not I酶切位点序列及保护碱基,上游引物引入Nhe I位点,下游引入Not I位点,引物序列由上海生工有限公司合成。扩增引物序列及酶切位点序列(见表1)。
表1 扩增引物序列及酶切位点序列
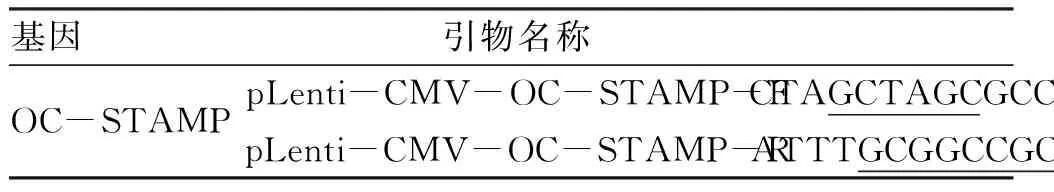
基因引物名称引物序列(5'-3')产物长度(bp)OC-STAMPpLenti-CMV-OC-STAMP-FCTAGCTAGCGCCACCATGAGGACCATCAGGGCAGCCACGGpLenti-CMV-OC-STAMP-RATTTGCGGCCGCCTACTCGAGGTCATATGGAGGCCCA 1496
划线部分为酶切位点,前面为保护碱基。
1.2.2 OC-STAMP基因的扩增质粒构建及鉴定 以小鼠单核细胞株RAW264.7细胞的总RNA反转录所得cDNA为模板,PCR扩增OC-STAMP基因序列目的片段(95 ℃ 5 min;94 ℃ 30 s,64 ℃ 40 s,72 ℃ 40 s为1个循环,共35个循环;72 ℃ 10 min)。PCR产物进行2%琼脂糖凝胶电泳,鉴定扩增产物,割胶回收试剂盒纯化回收后和pCDH-CMV载体都进行Nhe I/Not I双酶切,酶切产物经T4 DNA连接酶连接,构建为pLenti-CMV-OC-STAMP重组质粒。连接产物转化DH 5感受态细胞,37 ℃温箱孵育14 h,获得在氨苄抗生素筛选平板上生长的菌落; 挑取阳性克隆后4 mL LB液体培养基(含氨苄抗生素1∶1 000),180~220 rpm,37 ℃ 14 h,应用小提试剂盒抽取质粒(Takara),再经双酶切鉴定,将酶切大小正确的阳性质粒送上海生工有限公司测序分析,结果经Blast比对。
1.2.3 293T细胞转染及病毒包装 质粒验证构建成功后,将质粒转化DH 5感受态细胞,37 ℃温箱孵育14 h,获得在氨苄抗生素筛选平板上生长的菌落; 挑取阳性克隆后100 mL LB液体培养基(含氨苄抗生素1∶1 000),200~250 rpm,37 ℃ 14 h,应用大提试剂盒抽取质粒(天根)。质粒抽提成功后,利用Lipofectamine 3 000脂质体转染法转染293 T细胞,荧光显微镜下观察荧光判断转染程度。并用Western blot验证OC-STAMP过表达情况。采用两包装质粒系统(psPAX2/pMD2.G)包装目的质粒,以各质粒个数比(目的质粒:psPAX2∶pMD2.G=1∶1 ∶1)混合后用Lipofectamine 3 000共转染293 T细胞,分别收集48和72 h的病毒液,并用过滤器(Millipore)5 000 rpm×15 min过滤收集浓缩病毒液,于-80 ℃保存。
1.2.4 OC-STAMP过表达稳转细胞系的构建 将RAW 264.7细胞以1×105/孔的密度接种于6孔板中,待细胞铺板约50%~60%时换用病毒液1 mL(含Polybrene 8 μg/mL),培养约4~6 h后用低血清培养液opti-medium(Hyclone)补到2 mL,继续培养,第二天换液。继续培养约72 h后,加入Puromycin 2.5 μg/mL进行筛选3 d。最终获得的细胞继续传代培养,q-PCR和Western blot验证OC-STAMP稳定过表达。
1.2.5 荧光定量PCR Trizol法提取OC-STAMP过表达稳转系RAW 264.7细胞和对照组正常细胞的总RNA,RNA浓度通过Nanodrop光度计检测(260 nm/280 nm)。1 μg总量RNA采用TaKaRa反转录试剂盒进行RT反应,生成cDNA。SYBR Green荧光定量法检测OC-STAMP mRNA水平,反应体系20 μL(SYBR premix Ex TaqII 10 μL;PCR forward/reverse primer各0.8 μL;dH2O 6.4 μL;模板cDNA 2 μL)预变性95 ℃ 30 s;95 ℃ 5 s,60 ℃ 30 s为1个循环,共40循环。所用引物序列(见表2)。
表2 荧光定量PCR引物序列
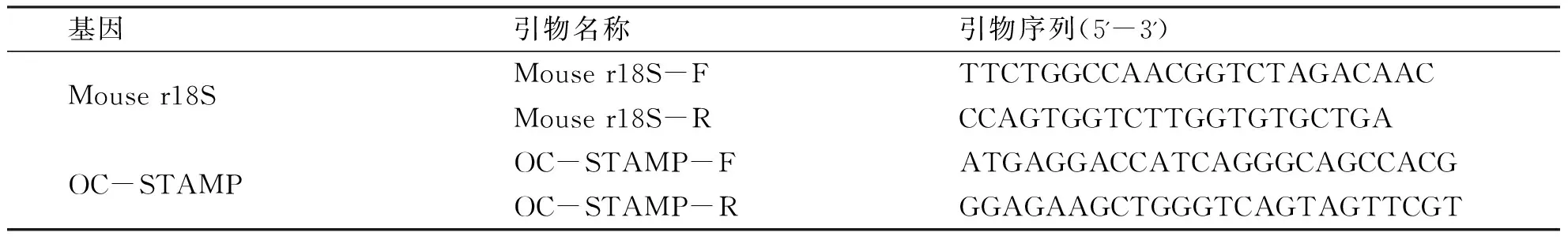
基因引物名称引物序列(5'-3')Mouser18SMouser18S-FTTCTGGCCAACGGTCTAGACAACMouser18S-RCCAGTGGTCTTGGTGTGCTGAOC-STAMPOC-STAMP-FATGAGGACCATCAGGGCAGCCACGOC-STAMP-RGGAGAAGCTGGGTCAGTAGTTCGT
1.2.6 Western blotting检测OC-STAMP 293 T细胞转染pCDH-CMV-oc-stamp重组质粒及pCDH-CMV-vector后48 h和过表达OC-STAMP稳转株RAW 264.7细胞及对照组细胞铺满皿约90%时,吸弃培养液,用预冷的PBS轻洗3遍,加入含有蛋白酶抑制PMSF-NaF的细胞裂解液RIPA(P 0013,Beyotime),收集蛋白,离心取上清,BCA(Beyotime)测定蛋白浓度。取适量蛋白加入loading buffer混匀,99 ℃加热5 min,等量上样,经SDS-PAGE电泳,并转至PVDF膜。5%脱脂奶粉封闭1~2 h,分别用OC-STAMP抗体(1 ∶500,Santa Cruz Biotechnology)和兔GAPDH抗体(1 ∶1 000,CST)4 ℃孵育过夜,TBST洗3次,每次5~10 min。分别用辣根过氧化物酶标记的山羊抗兔(1∶5 000,Jackson)二抗室温孵育1 h,TBST洗膜,添加ECL化学发光显色液,显影读片。
2 结果
2.1 重组pCDH-CMV/OC-STAMP质粒的鉴定 将构建好的pCDH-CMV/OC-STAMP重组质粒用NehI及NotI进行双酶切,酶切产物经琼脂糖凝胶电泳后出现的条带与预测的大小相一致(见图1 A),进一步经测序证实所构建的小鼠OC-STAMP蛋白表达载体序列正确,框架基本无误(见图1 B)。

A:重组质粒的双酶切鉴定;B:重组质粒测序的部分序列。1:为重组质粒未用限制性内切酶酶切;2:为重组质粒双酶切后。图1 pLenti-CMV-OC-STAMP重组质粒的双酶切及测序
2.2 重组质粒转染293T细胞后的荧光表达及OC-STAMP蛋白表达 将成功构建的pCDH-CMV/OC-STAMP重组质粒以及对照vector质粒转染293 T细胞48 h后可见大量绿色荧光,提示质粒成功转染,并表达GFP(见图2 A)。然后收集细胞,用RIPA裂解提取总蛋白,Western blot结果显示:转染pCDH-CMV/OC-STAMP重组质粒后,293 T细胞中OC-STAMP表达量显著增高,且差异具有统计学意义(见表3和图2 B)。进一步证明重组质粒的构建成功。

A:对照空载质粒和重组质粒转染293 T细胞48 h后,荧光显微镜下观察GFP的表达(×100,图中标尺为100 μm)。B:对照空载质粒和重组质粒转染293 T细胞后,OC-STAMP的蛋白表达。*:P<0.05。图2 重组质粒转染293T细胞后的荧光GFP表达及OC-STAMP的蛋白表达


蛋白组别面积平均灰度值累计光密度GAPDH空载对照2728.00±35.03197.02±3.38537387±3453.80质粒转染2834.30±27.10192.79±0.19546361±1737.15OC-STAMP空载对照3546.00±40.36150.78±1.56534631±587.73 质粒转染7269.67±49.16189.17±1.901375188±6215.01
2.3 荧光定量PCR和Western blot验证稳转RAW264.7细胞后OC-STAMP的表达 采用psPAX2/pMD2.G两包装质粒系统包装目的质粒获得病毒液后,感染RAW 264.7细胞,经嘌呤霉素筛选后传代培养获得表达OC-STAMP的稳转株细胞。Trizol法提取总RNA,进行逆转录反应生成cDNA,并进行荧光定量PCR检测。如图3 A和表4所示,与感染对照组细胞相比,过表达感染组细胞中的OC-STAMP mRNA水平显著提高,并且具有统计学意义。同时采用RIPA裂解液裂解稳转株细胞提取细胞总蛋白,Western blot结果显示:过表达OC-STAMP的稳转株细胞系成功过表达了OC-STAMP蛋白含量,较空白对照组增加了约2.5倍(见表5和图3 B)。以上结果显示OC-STAMP的过表达稳转细胞系构建成功。
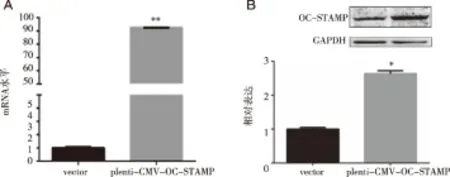
A:重组质粒稳转RAW 264.7细胞后OC-STAMP的mRNA水平;B:重组质粒稳转RAW264.7细胞后OC-STAMP的蛋白水平。*:P<0.05;**:P<0.01。图3 重组质粒稳转RAW 264.7细胞后OC-STAMP的表达
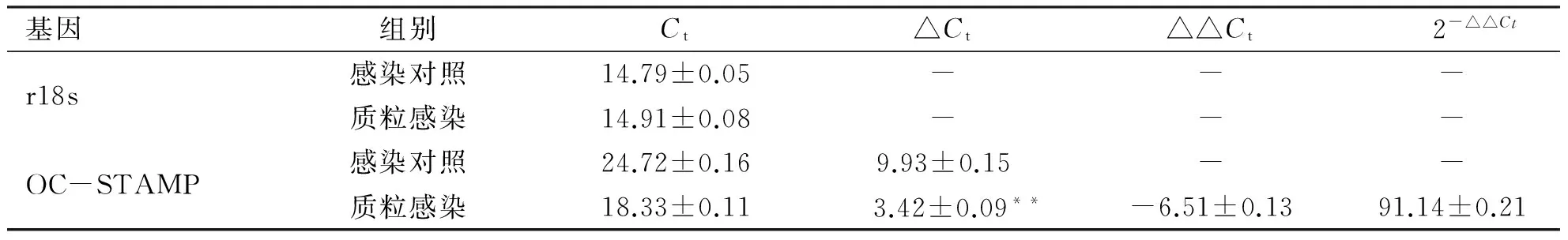
基因组别Ct△Ct△△Ct2-△△Ctr18s感染对照14.79±0.05---质粒感染14.91±0.08---OC-STAMP感染对照24.72±0.169.93±0.15--质粒感染18.33±0.11 3.42±0.09**-6.51±0.1391.14±0.21
与感染对照组相比,**P<0.01。
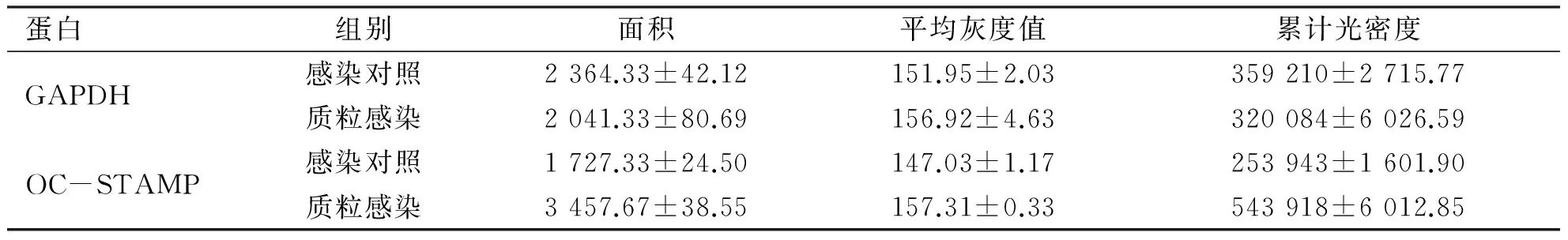
蛋白组别面积平均灰度值累计光密度GAPDH感染对照2364.33±42.12151.95±2.03359210±2715.77质粒感染2041.33±80.69156.92±4.63320084±6026.59OC-STAMP感染对照1727.33±24.50147.03±1.17253943±1601.90质粒感染3457.67±38.55157.31±0.33543918±6012.85
3 讨论
OC-STAMP基因的功能尚不明确。以往研究表明,OC-STAMP在经RANKL诱导后表达量可明显上调,促进破骨细胞分化和融合,且呈剂量依赖性[1]。Kim等[2]在RANKL诱导下的RAW 264.7和原代骨髓巨噬细胞中,分别加入PKCβ、Akt、P38、NF-κB、c-Jun氨基末端激酶(JNK)等的阻断剂均可引起OC-STAMP mRNA表达水平的下调。同样是在小鼠巨噬细胞株RAW 264.7及原代骨髓巨噬细胞中,氧化低密度脂蛋白可通过抑制ERK、P38、JNK和转录因子NF-κB、NFAT的活性来阻断RANKL信号通路,从而抑制破骨细胞分化,且这种抑制作用呈剂量依赖性[18]。高浓度右旋葡萄糖则能通过抑制细胞内ROS的释放抑制NF-κB活性,还能抑制RANKL诱导的破骨细胞分化及趋化运动[19]。致炎因子白介素17可直接刺激多种细胞(巨噬细胞、内皮细胞、上皮细胞等)分泌RANKL,同时还可以通过刺激细胞合成分泌IL-1、L-6等促进细胞分泌RANKL,诱导破骨细胞形成,促进骨吸收[20]。
RANKL是目前发现的惟一具有诱导破骨细胞分化发育发挥功能的因子,与NF-κB受体活化因子(receptoractivatorof nuclearfactor-B,RANK)结合后可引起RANK结构的改变,激活位于RANK胞质侧的肿瘤坏死因子受体连接蛋白6(TRAF 6),形成RANK-TRAF 6受体复合物,进而通过招募下游信号因子(PKC、NFAT、P38、JNK及NF-κB等)激活NF-κB、MAPK、PI3K/Akt等通路[21],形成RANKL-RANK轴,促进巨噬细胞的分化成熟,抑制凋亡维持存活,影响巨噬细胞的分化移行[18-19,21]。
本实验从小鼠单核巨噬细胞RAW 264.7中提取总RNA,经反转录、聚合酶链反应得到小鼠OC-STAMP的全长序列,克隆至pCDH-CMV的慢病毒载体中,双酶切及基因测序表明重组质粒构建成功,与NCBI基因库中的鼠OC-STAMP碱基序列基本符合。将重组质粒转染至293 T细胞,荧光显微镜下观察到大量荧光,提示具有良好的转染效率和表达效率。最后,提取过表达组及对照组细胞的总蛋白,经Western blot进一步证实过表达重组质粒的构建成功。质粒的成功构建为深入了解OC-STAMP的蛋白功能提供了有效工具。
此后,我们进一步构建了过表达OC-STAMP的稳转细胞系。Real-time PCR和Western blot的结果显示,RAW 264.7稳转细胞较好地过表达了OC-STAMP。本实验最终获得的稳转细胞系是混合细胞系,后期可能会存在细胞的过表达效应降低。由于实验技术能力局限,未能将该稳转系成功单克隆化。但我们会继续探索,尽可能将该稳转细胞系单克隆化。
膜蛋白在免疫反应中扮演着非常重要的作用,他们与相应的配体(如炎症因子、生长因子、趋化因子等)结合,激活下游信号通路,调控相关基因的表达。基于已有研究结果和我们的前期工作,我们设想OC-STAMP参与了RANKL-RANK轴引发的巨噬细胞相关的反应,有可能在糖尿病状态下的大血管病变进程中扮演某种角色。在现有的研究基础上应用慢病毒携带的过表达技术和沉默技术以及抗体中和技术,观察OC-STAMP对于巨噬细胞的调控作用及潜在机制,并寻找其可能配体。本实验构建的OC-STAMP过表达载体及过表达稳转株细胞的成功制备将有助于对OC-STAMP在巨噬细胞中信号通路的调控机制展开更深入的研究。
[1] Yang M,Birnbaum M J,MacKay C A,et al.Osteoclast stimulatory transmembrane protein (OC-STAMP),a novel protein induced by RANKL that promotes osteoclast differentiation[J].J Cell Physiol,2008,215(2):497-505.
[2] Kim M H,Park M,Baek S H,et al.Molecules and signaling pathways involved in the expression of OC-STAMP during osteoclastogenesis[J].Amino Acids,2011,40(5):1447-1459.
[3] Takagi T,Inoue H,Takahashi N,et al.Sulforaphane inhibits osteoclast differentiation by suppressing the cell-cell fusion molecules DC-STAMP and OC-STAMP[J].Biochem Biophys Res Commun,2017 ,483(1):718-724.
[4] Sambandam Y,Baird K L,Stroebel M,et al.Microgravity induction of TRAIL expression in preosteoclast cells enhances osteoclast differentiation[J].Sci Rep,2016,6:25143.
[5] Cheon Y H,Kim J Y,Baek J M,et al.WHI-131 promotes osteoblast differentiation and prevents osteoclast formation and resorption in Mice[J].J Bone Miner Res,2016,31(2):403-415.
[6] Dou C,Li J,Kang F,et al.Dual effect of cyanidin on RANKL-induced differentiation and fusion of osteoclasts[J].J Cell Physiol,2016,231(3):558-567.
[7] Kiyomiya H,Ariyoshi W,Okinaga T,et al.IL-33 inhibits RANKL-induced osteoclast formation through the regulation of Blimp-1 and IRF-8 expression[J].Biochem Biophys Res Commun,2015,460(2):320-326.
[8] Witwicka H,Hwang S Y,Reyes-Gutierrez P,et al.Studies of OC-STAMP in osteoclast fusion:a new knockout mouse model,rescue of cell fusion,and transmembrane topology[J].Plos One,2015,10(6):e0128275.
[9] Kang M R,Jo S A,Yoon Y D,et al.Agelasine D suppresses RANKL-induced osteoclastogenesis via down-regulation of c-Fos,NFATc1 and NF-κB[J].Mar Drugs,2014,12(11):5643-5656.
[10] Hwang Y S,Ma G T,Park K K,et al.Lysophosphatidic acid stimulates osteoclast fusion through OC-STAMP and P2X7 receptor signaling[J].J Bone Miner Metab,2014,32(2):110-122.
[11] Miyamoto T.STATs and macrophage fusion[J].JAKSTAT,2013,2(3):e24777.
[12] Khan U A,Hashimi S M,Bakr M M,et al.Foreign body giant cells and osteoclasts are TRAP positive,have podosome-belts and both require OC-STAMP for cell fusion[J].J Cell Biochem,2013,114(8):1772-1778.
[13] Xing L,Xiu Y,Boyce B F.Osteoclast fusion and regulation by RANKL-dependent and independent factors[J].World J Orthop,2012,3(12):212-222.
[14] Miyamoto H,Suzuki T,Miyauchi Y,et al.Osteoclast stimulatory transmembrane protein and dendritic cell-specific transmembrane protein cooperatively modulate cell-cell fusion to form osteoclasts and foreign body giant cells[J].J Bone Miner Res,2012,27(6):1289-1297.
[15] Miyamoto H,Katsuyama E,Miyauchi Y,et al.An essential role for STAT6-STAT1 protein signaling in promoting macrophage cell-cell fusion[J].J Biol Chem,2012,287(39):32479-32484.
[16] Oursler M J.Recent advances in understanding the mechanisms of osteoclast precursor fusion[J].J Cell Biochem,2010,110(5):1058-1062.
[17] 胡其娴,马晓文,胡小磊,等.小鼠OC-STAMP蛋白表达纯化抗体制备及鉴定[J].中华临床医师杂志:电子版,2013,7(8):3412-3415.
[18] Mazière C,Louvet L,Gomila C,et al.Oxidized low density lipoprotein decreases Rankl-induced differentiation of osteoclasts by inhibition of Rankl signaling[J].J Cell Physiol,2009,221(3):572-578.
[19] Wittrant Y,Gorin Y,Woodruff K,et al.High d(+) glucose concentration inhibits RANKL-induced osteoclastogenesis[J].Bone,2008,42(6):1122-1130.
[20] 杨学辉.白细胞介素-17在骨代谢与炎症性骨疾病的作用[J].遵义医学院学报,2016,39(5):441-448.
[21] Boyle W J,Simonet W S,Lacey D L.Osteoclast differentiation andactivation[J].Nature,2003,423(6937):337-342.
[收稿2017-02-10;修回2017-03-22]
(编辑:王静)
Construction of the OC-STAMP over-expressed vector and its stable expression cell strain
YuanHuimin1,HeJiangping2,ZhangGuangya2,ZhangDandan2,ChenFengling2
(1.Clinical College,Bengbu Medical College,Bengbu Anhui 233030,China;2.Department of Endocrinology,Shanghai Ninth People’s Hospital,School of Medicine of Shanghai Jiao Tong University,Shanghai 201900,China)
Objective To construct the Osteoclast stimulatory transmembrane protein (OC-STAMP) over-expressed plasmid and transfect RAW264.7 cell in vitro to construct stable expression cell strain.Methods Primers were designed according to the gene information on OC-STAMP in National Center for Biotechnology Information,and the OC-STAMP gene was amplified by Polymerase Chain Reaction (PCR).Using the gene recombination technology,the double enzyme-digested OC-STAMP gene was cloned into the pCDH-CMV-MCS-EF1-Puro vector with GFP.Recombined plasmids were identified with enzyme digestion and sequencing and transfected into 293T cell,.Western blotting was used to detect its expression.Lentiviruses were harvested using pLenti-CMV-OC-STAMP co-transfected with psPAX2 and pMD2.G into HEK293T cells,then RAW264.7 cells were transduced with lentiviruses.Quantitative real-time RT-PCR and western blotting were used to confirm its over-expression.Results The results of enzyme digestion and sequencing revealed that the recombinant plasmids pLenti-CMV-OC-STAMP was successfully constructed.After transfecting 293T cells with the successfully constructed plasmid pLenti-CMV-OC-STAMP,a largeamount of green fluorescence was observed.After transfecting RAW264.7 cells with lentiviruses,the results of quantitative real-time RT-PCR and western blotting showed that the successful over-expression of pLenti-CMV-OC-STAMP.Conclusion The recombinant plasmids pLenti-CMV-OC-STAMP is successfully constructed and the stable expression cell strain is also constructed successfully.
osteoclast stimulatory transmembrane protein; plasmid construction; lentiviruses transfection; RAW264.7 cells.
上海市宝山区科学技术委员会科研项目(NO:14-E-3);上海交通大学医学院博士创新基金资助项目(NO:BXJ201638)。
陈凤玲,女,博士,主任医师,博士生导师,研究方向:糖尿病大血管病变发病机制及干预、线粒体糖尿病的基因突变与糖脂代谢紊乱,E-mail:cfl1993@126.com。
R589.9
A
1000-2715(2017)02-0143-07
