高效液相色谱-示差折光检测法测定柴油中的芳烃含量
2017-06-08马宏园安晓春江明玉刘毅
马宏园,安晓春,江明玉,刘毅
(1.广东省茂名市质量计量监督检测所,广东 茂名 525000;2.中国石油化工股份有限公司茂名分公司质量检验中心,广东 茂名 525011;3.重庆出入境检验检疫局检验检疫技术中心,重庆 400020)
高效液相色谱-示差折光检测法测定柴油中的芳烃含量
马宏园1,安晓春1,江明玉2,刘毅3
(1.广东省茂名市质量计量监督检测所,广东 茂名 525000;2.中国石油化工股份有限公司茂名分公司质量检验中心,广东 茂名 525011;3.重庆出入境检验检疫局检验检疫技术中心,重庆 400020)
采用高效液相色谱-示差折光检测法测定柴油芳烃含量,氨基柱作固定相,正庚烷为流动相,辅以反冲技术将样品分离成非芳烃、单环芳烃、双环芳烃和三环+芳烃等组分。考察流动相流量、色谱柱温度、进样量、反冲时间等条件对结果的影响。方法的相对标准偏差在0.9%~6.1%范围内,回收率在91.5%~109.1%之间。该方法快速、简便,可在22min内完成一个柴油样品芳烃含量的测定,能很好地满足柴油芳烃快速检测的要求。
柴油;芳烃;高效液相色谱;示差折光检测;反冲技术
0 引言
随着柴油中芳烃尤其是多环芳烃含量的增加,运输工具尾气排放物中的固体颗粒物、氮氧化物的含量也相应增加,因而柴油中的芳烃含量逐渐受到人们的关注。美国和欧盟对柴油中多环芳烃含量进行了限制;我国也在不断完善柴油的质量标准,2004年,北京市政府在地方标准[1]中规定了多环芳烃含量的限值;2006年,广州市颁布的车用柴油地方标准[2]中也加入了多环芳烃含量的限值。2013年全国开始实施的车用柴油强制性国家标准[3]中,也明确规定了芳烃含量限值,因此准确测定柴油中的芳烃含量非常重要。
目前测定柴油中芳烃含量的方法较多,主要有荧光指示剂色谱法(FIA)[4]、色谱-质谱联用法(LC-MS)[5]、高效液相色谱-示差折光检测器法(HPLC-RID)[6]、超临界流体色谱法(SFC)[7]等,以上方法各有优缺点,FIA测定样品的终馏点不能大于315℃,也不能测定有色样品,但目前炼厂柴油终馏点一般在360℃左右,且大部分呈浅黄色,显然该法不合适;LC-MS法虽能得到准确、详细的族组成信息,但它需先将柴油分离成饱和烃与芳烃,费时费力,不太适合常规的出厂检测;SFC法由于受仪器、试剂等条件的限制,使用还有一定困难。
HPLC-RID法虽然由于多环芳烃组分的校正因子问题对测定结果有一定影响[8],但该法简便、快速并且有良好的重复性和准确性[9],已被制定成石化行业标准SH/T 0806——2008[6],并在新的柴油国家标准中被引用成为常规检测方法。本文采用上述标准方法对柴油中的单环芳烃、双环芳烃和三环+芳烃等组分进行分离和测试,确定最佳测定条件,使用标准曲线法定量,结果满意。
1 试验部分
1.1 主要仪器和试剂
Agilent 1200型高效液相色谱仪,配G1322A型真空脱气机、G1362A型示差折光检测器、7725i手动进样器、ChemStation色谱工作站,美国安捷伦公司;有机相针式过滤器,13mm×0.45μm,上海安谱科学仪器有限公司。
正庚烷,HPLC级,美国TEDIA公司;环己烷,质量分数>99.5%,上海试剂一厂;邻二甲苯、1-甲基萘,质量分数>99%,国药集团化学试剂有限公司;菲、二苯并噻吩,质量分数>98%,美国J&K CHEMICAL公司;9-甲基蒽,质量分数>99%,比利时ACROS公司。
1.2 色谱条件
色谱柱:安捷伦公司ZORBAX NH2柱(250mm× 4.6mm,5μm),配氨基保护柱(12.5mm×4.6mm,5μm);流动相:正庚烷,流量0.8mL/min;柱温:35℃;检测器温度:35℃;进样量:20μL。
1.3 标准溶液的配制
1.3.1 系统性能验证标准溶液(SPS)
称量(1.0±0.1)g环己烷、(0.5±0.05)g邻二甲苯、(0.05±0.005)g二苯并噻吩、(0.05±0.005)g 9-甲基蒽,置于同一100mL容量瓶中,用正庚烷稀释至刻度。
1.3.2 标准溶液
称量各标准物质(准确到0.000 1 g)置于100mL容量瓶中,用正庚烷稀释至刻度,按照表1所述质量浓度系列配制标准溶液A,B,C,D。

表1 标准溶液的配制g/100mL
1.4 实验步骤
将柴油试样先用正庚烷稀释,定量注入HPLC系统中,非芳烃、单环芳烃、双环芳烃和三环+芳烃在极性柱中依次流出。双环芳烃流出以后,在一预先测定的反冲点,让柱子反冲洗,将三环+芳烃洗脱成一尖锐的窄峰。利用示差折光检测器(RID)检测,外标标准曲线法定量。
2 结果与讨论
2.1 色谱条件的选择
2.1.1 色谱柱的选择
柴油中烷烃属于非极性物质,芳烃属于弱极性物质,因此,使用正向、反向色谱在理论上都是可行的,但考虑到样品需要使用有机溶剂溶解,流动相最好使用有机溶剂,如正庚烷,因此与之配套的色谱柱为极性柱,氨基柱与氰基柱都可使用,本方法只对氨基柱进行了考察。
实验中发现,样品含有微量极性化合物,氨基柱对极性物有不可逆吸附,吸附在柱头的极性物用正庚烷很难冲出。为维持柱效,延长柱的使用寿命,通过在氨基柱前加氨基保护柱,减少污染,有效地保护分析柱。
2.1.2 流动相流量的选择
在本文方法中,系统的分辨率(分离度)定义为:2(t2-t1)/(y1+y2),其中t2、t1分别为环己烷、邻二甲苯的保留时间,y1、y2分别为环己烷、邻二甲苯的峰宽;系统的反冲时间定义为:ta+0.4(tb-ta),其中ta、tb分别为二苯并噻吩、9-甲基蒽的保留时间,反冲时间能反映系统出峰速度(特别是双环、三环芳烃)的快慢。
固定其他条件,考察流动相不同流量对系统分辨率和反冲时间的影响,实验结果如表2所示。
从表中可以看出,随着流量的增加,系统的分辨率下降,分离能力下降,但是反冲时间减小,也就是出峰速度加快。分析方法需要在保证分辨率的前提下尽量减少分析时间,以提高分析速度。本方法选择流量为0.8mL/min。
2.1.3 柱温的选择
柱温的变化对样品中各组分的保留值及分离度有一定的影响,当温度波动范围较大时,会影响定量结果的准确性和重复性。在其他条件固定的情况下,考察不同柱温下系统反冲时间及分辨率的变化。

表2 流量对系统分辨率和反冲时间的影响
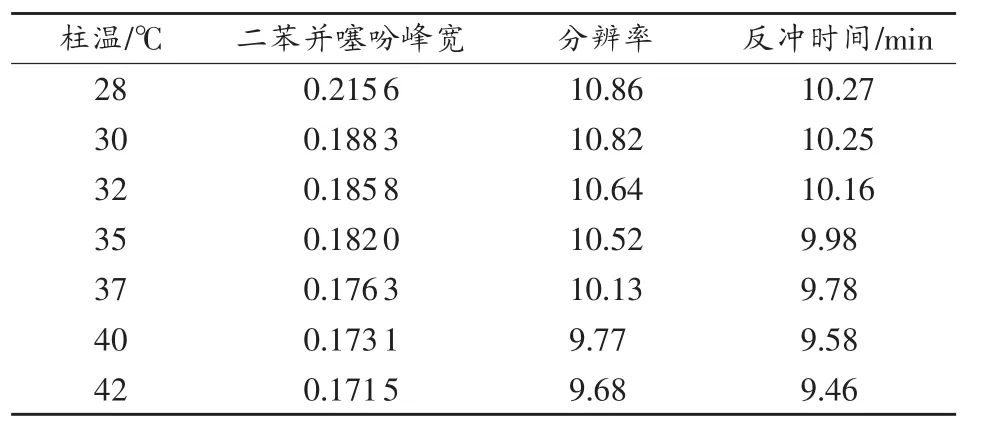
表3 柱温对系统分辨率和反冲时间的影响
从表中可以看出,当柱温较低时,尽管各类组分间的分离度增大,但此时多环芳烃谱峰的半峰宽较大,峰形变差;随柱温升高,分析时间缩短,但饱和烃与单环芳烃间的分离度变差,综合考虑这两种情况,选择柱温为35℃。
2.1.4 检测器温度的选择
介质的密度受温度的影响最大,因此,示差折光检测器对温度的变化极为敏感,检测灵敏度和最小检出量常取决于温度控制精度,需要将精度控制在±0.001℃。一般来说,检测器的温度至少高于环境5℃才能很好地控制温度恒定,室温通常为26℃左右,因此检测器温度设定为30℃以上并保持恒定。
实验发现,不同恒定的检测器温度对系统的分辨率以及反冲时间无明显的影响,根据检测器温度与柱温设置尽量相同的原则,选择检测器温度为35℃。
2.2 系统反冲时间的确认
柴油组成复杂,芳环的多少和排列方式、取代数目及取代位置等因素都对芳烃的保留时间产生影响,造成不同环数的芳烃在色谱图上的部分重叠,从而影响到芳烃的准确定量,因此确定色谱图中各类芳烃的切割位置对芳烃含量的准确测定非常重要。
从SPS的色谱图1可以看出,在本方法的分离条件下,非芳烃、单环芳烃能较早洗出,分辨率也很好,但双环、三环+较迟洗出,且分辨率不高。对于柴油样品,从色谱图2可以看出双环、三环+的界限更为模糊。
SH/T 0806——2008中对柴油各种芳烃是以保留时间的相对大小来定义的,方法规定在双环芳烃流出后对柱子进行反冲洗,后洗出的峰为三环+芳烃,这个反冲时间由SPS的谱图计算得到。已有研究者利用光电二极管阵列检测器(PDAD)与示差折光检测器(RID)串联系统对方法反冲时间允差值进行了详细考察[10],结果表明,其允许变化范围为±0.2min。
在其他条件固定的情况下,利用同一SPS考察日内与日间反冲时间的变化,其结果见图3和图4。

图1 典型SPS色谱图
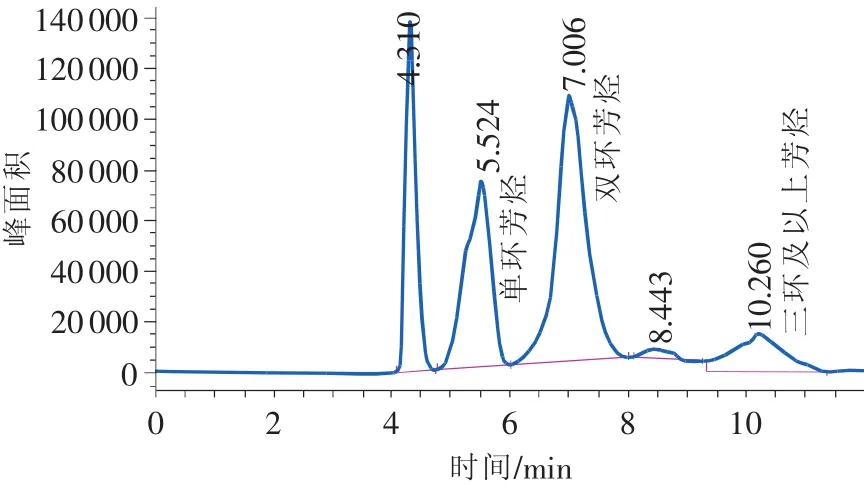
图2 未反冲的柴油样品色谱图
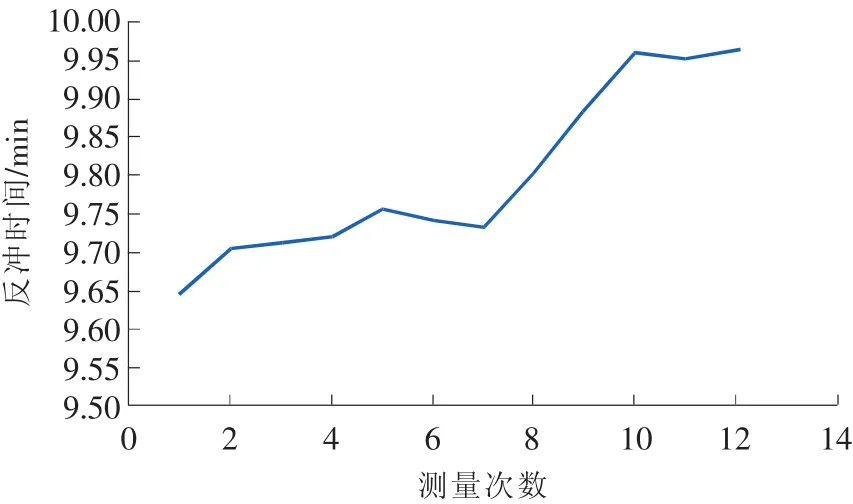
图3 同一日内间隔30min反冲时间变化图
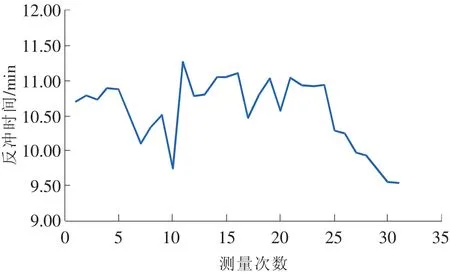
图4 连续30个工作日反冲时间变化图
从图中可以看出,不管是日内还是日间,其反冲时间的变化都超过了0.2min。因此,测定样品时必须每天用SPS测定反冲时间,不能沿用旧反冲时间。最好在利用SPS确定反冲时间后马上进行样品的测定,减少因反冲时间波动而引起的测定误差。
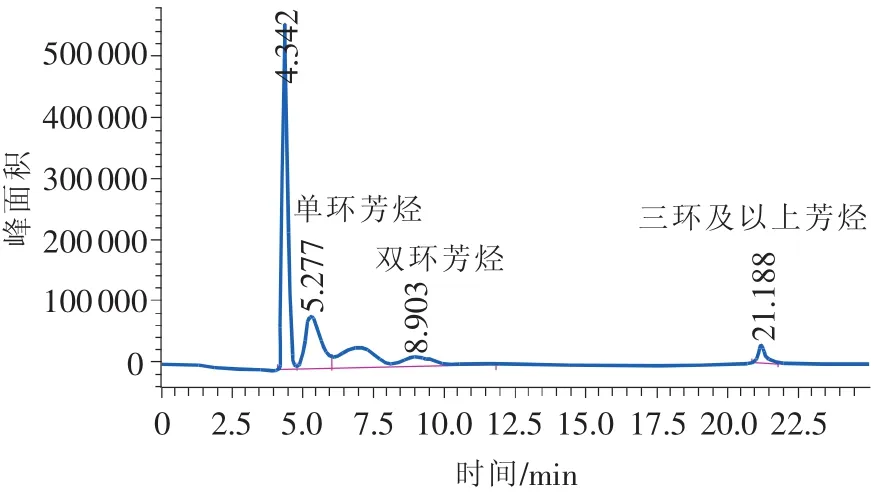
图5 直馏柴油样品色谱图

图6 催化柴油样品色谱图

图7 加氢柴油样品色谱图
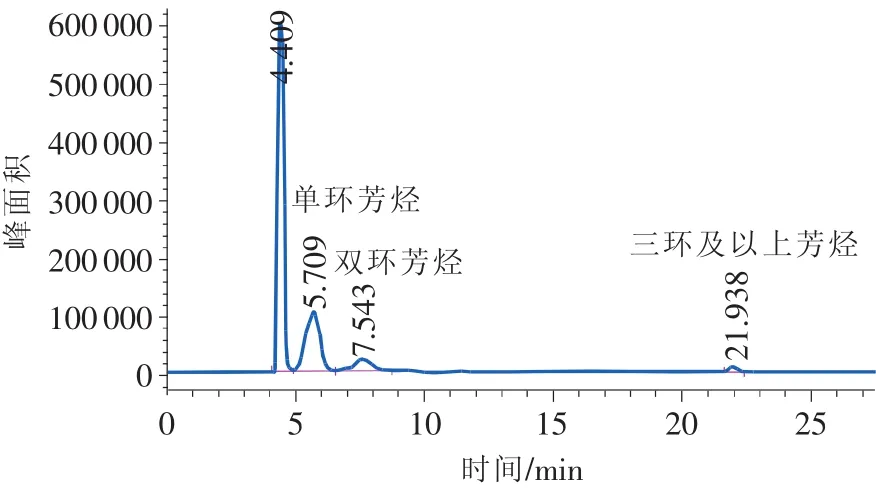
图8 0#柴油样品色谱图

图9 -10#军用柴油样品色谱图
试验发现,反冲时间一般为10min左右,在22min以内能够完成一个样品的分析。
典型的柴油分析色谱图如图5~图9所示。
2.3 样品处理与进样量的选择
样品处理方法为:称取0.9~1.1g的样品,用正庚烷稀释到10mL,混匀定容,进样前使用0.45μm的有机滤膜过滤样品。对于检测成分含量不在工作曲线范围的样品,要加大或减少称样量。
在保证合理检测限的前提下,避免柱超载是选择进样量的首要考虑问题。在5,10μL的进样量下,SPS谱图中的二苯并噻吩及9-甲基蒽信噪比小于3∶1,出峰不明显;在30μL的进样量下,样品峰出现拖尾,表明柱超载,因此选择20μL的进样量。本系统采用7725i手动进样器,配套20μL定量环全充满方式进行定量进样。
2.4 标准曲线
按照1.3.2的方法配制标准溶液,在选定的试验条件下,进行标样分析,得出的回归方程为:单环芳烃:y=2 220 600x+849,r=0.999 999 61;双环芳烃:y= 3 842 900x+13 994,r=0.999 997 49;三环+芳烃:y= 4 659 400x+594,r=0.999 988 19;其中y值为峰面积,x值为质量浓度。可以看出,各芳烃曲线具有良好的线性,完全能够满足SH/T 0806——2008标准中规定的相关系数要大于0.999,截距要小于0.01g/100mL的要求。

表4 精密度试验结果1)%(质量分数)

表5 加标回收的试验结果
2.5 精密度和准确度
在选定的条件下,重复测定柴油样品,计算测定结果及相对标准偏差(RSD),结果见表4。对同一样品测定10次的相对标准偏差(RSD)在0.9%~6.1%,满足色谱分析的精密度要求,且同一样品多次测定的极差均低于SH/T 0806——2008的允差。
在样品中加入一定量的芳烃标准物质,测定回收率以检验方法的准确度,试验结果见表5。加标回收率在91.5%~109.1%之间,表明本方法准确度较高。
3 结束语
本文采用液相色谱-示差折光检测法测定柴油中芳烃的含量,固定相为氨基键合硅胶;流动相为正庚烷,流量0.8mL/min;柱温35℃;示差检测器温度35℃;进样量20μL。方法的相对标准偏差在0.9%~6.1%范围内,回收率在91.5%~109.1%之间,具有较好的精密度和准确度。方法快速、简便,可在22min内完成测定,能够很好地满足柴油芳烃快速检测的要求。反冲时间是方法的一个重要参数,必须在每次测定样品前测定反冲时间,否则会引起测定误差。
[1]车用柴油:DB 11/239-2004[S].北京:中国标准出版社,2004.
[2]车用柴油:DB 44/346-2006[S].北京:中国标准出版社,2006.
[3]车用柴油:GB 19147-2013[S].北京:中国质检出版社,2013.
[4]液体石油产品烃类的测定(荧光指示剂吸附法):GB/T 11132-2008[S].北京:中国标准出版社,2008.
[5]中间馏分烃类组成测定法(质谱法):SH/T 0606-2005[S].北京:中国石化出版社,2006.
[6]中间馏分芳烃含量的测定(示差折光检测器高效液相色谱法):SH/T 0806-2008[S].北京:中国石化出版社,2008.
[7]Determination of the aromatic content and polynuclear aromatic content of diesel fuels and aviation turbine fuels by supercritical fluid chromatography:ASTM D5186-2003[S].2003.
[8]张大伟,祝馨怡,田松柏,等.响应因子对高效液相色谱法测定柴油族组成的影响[J].石油与天然气化工,2007,36(2):153-156.
[9]白正伟,李怿,林玉,等.柴油芳烃含量测定方法评价[J].石油商技,2009(3):86-91.
[10]林玉,李怿,白正伟,等.高效液相色谱-光电二极管阵列检测法测定柴油中芳烃的类型[J].色谱,2008,26(2):250-253.
(编辑:莫婕)
Determ ination of aromatic hydrocarbon content in diesel oil by HPLC-RID
MA Hongyuan1,AN Xiaochun1,JIANG Mingyu2,LIU Yi3
(1.Maoming Quality&Metrology Supervision Testing Institute,Maoming 525000,China;2.Quality Tested Tenter of SINOPEC Maoming Company,Maoming 525011,China;3.Inspection&Quarantine Technical Center of Chongqing Entry-Exit Inspection&Quarantine Bureau,Chongqing 400020,China)
HPLC-RID method is applied for the determination of aromatic hydrocarbon content in diesel oil,with amino column as stationary phase and n-heptane as mobile phase,supplemented with back-flushing technique for separating samples into components such as non-aromatic hydrocarbon,monocyclic aromatic hydrocarbon,bicyclic aromatic hydrocarbon and tricyclic+aromatic hydrocarbon.The effects of mobile phase flow rate,chromatographic column temperature,sample volume,back flushing time on the results are assessed.The relative standard deviation of the method is within the range of 0.9%-6.1%and the recovery rate is between 91.5%and 109.1%.The method is fast and simple,and can measure the aromatic hydrocarbon content in a diesel oil sample in 22 minutes,satisfying the requirement of fast detection of aromatic hydrocarbon content in diesel oil.
diesel oil;aromatic hydrocarbons;HPLC;RID;back-flushing technique
A
1674-5124(2017)05-0040-05
10.11857/j.issn.1674-5124.2017.05.009
2016-07-29;
2016-09-05
马宏园(1978-),男,湖北当阳市人,高级工程师,主要从事石油化工产品分析检验工作。
