苯二氮䓬类药物的药理学
2017-06-05司天梅刘铁桥张朝辉贾福军郭万军王高华王传跃牛雅娟刘铁榜向小军敏0
司天梅,刘铁桥,张朝辉,贾福军,郭万军,王高华,王传跃,牛雅娟,刘铁榜,向小军,赵 敏0
(1. 北京大学第六医院,北京市100191;2. 中南大学湘雅二医院精神卫生研究所,长沙市410011;3. 新乡医学院第二附属医院,新乡市453002;4. 广东省人民医院,广州市510030;5. 四川大学华西医院,成都市610041;6. 武汉大学人民医院,武汉市430060;7. 首都医科大学附属北京安定医院,北京市100088;8. 北京回龙观医院,北京市100096;9. 深圳市精神卫生中心,深圳市518020;10. 上海精神卫生中心,上海市200030)
·专论述评·
苯二氮䓬类药物的药理学
司天梅1,刘铁桥2,张朝辉3,贾福军4,郭万军5,王高华6,王传跃7,牛雅娟8,刘铁榜9,向小军2,赵 敏10
(1. 北京大学第六医院,北京市100191;2. 中南大学湘雅二医院精神卫生研究所,长沙市410011;3. 新乡医学院第二附属医院,新乡市453002;4. 广东省人民医院,广州市510030;5. 四川大学华西医院,成都市610041;6. 武汉大学人民医院,武汉市430060;7. 首都医科大学附属北京安定医院,北京市100088;8. 北京回龙观医院,北京市100096;9. 深圳市精神卫生中心,深圳市518020;10. 上海精神卫生中心,上海市200030)
苯二氮䓬类药物;药理学;脂溶性;药代动力学;禁忌证
1 前言
苯二氮䓬类药物(benzodiazepines,BZDs)发展至今已有300多种,其中50余种在临床中使用。BZDs的基本结构由一个含7个原子的二氮䓬环和两个苯环并联而成,多数BZDs的两个氮原子在第1和第4位,故被称为1,4-苯二氮䓬[1]。通过对其R1位的结构改变,衍生出了药理特点各不相同的BZDs,在临床上用于治疗焦虑、失眠、激越、癫痫发作、肌肉痉挛、酒依赖或口腔科手术前用药等。本文主要介绍BZDs的药理学特征以及临床常用的BZDs。
2 药理学特征
2.1 药理特性及机制
对BZDs药理机制的认识始于1963年合成的地西泮。1967年有研究者发现地西泮可加强γ-氨基丁酸(GABA)的抑制作用,并且依赖于内源性GABA的存在。10年后,几个研究组均在大脑γ-氨基丁酸A(GABAA)受体上发现了BZD受体,一个50kDa的膜蛋白。
GABAA受体是一种与氯离子通道偶联的配体门控离子通道受体,由3个亚单位α、β和γ以2:2:1比例组成,围成一个玫瑰花形,中间为氯离子通道。BZD受体是GABAA受体上的一个异构性调节位点,这个复合型受体被称为BZD-GABAA受体复合物,BZDs是BZDGABAA受体复合物的非选择性完全激动剂。BZDs不直接激活GABAA受体,通过增强GABA与GABAA受体的效应发挥其作用。
GABAA受体的α亚单位有六种亚型(α1、α2、α3、α4、α5和α6),亚单位α正是BZD受体所在区域,不同的α亚单位及其与BZDs药物的亲和性高低,决定了BZDs的药理特性。由α1、α2、α3或α 5亚单位构成的GABAA受体,与BZDs亲和性较高,被称为BZDs敏感型GABAA受体;由α4或α6亚单位构成的GABAA受体与BZDs亲和性较低,被称为BZDs不敏感型GABAA受体[2,3]。
目前发现的BZDs受体包括两种中枢受体,BZD1(ω1或Ω1)和BZD2(ω2或Ω2),以及一种外周BZD受体(peripheral benzodiazepine receptor, PBR)。BZD1受体主要由α1亚单位构成,广泛分布于中枢神经系统,特别是大脑皮质、丘脑和小脑等网状结构,主要介导BZDs的抗焦虑、镇静和抗惊厥作用。与BZD1(ω1)受体高亲和性的BZDs,具有显著镇静、催眠和抗惊厥作用,可能会有顺行性遗忘和依赖的倾向[4,5]。BZD2受体是一类异质性受体,由α2或α3或α 5亚单位构成,主要分布于大脑皮层、海马、纹状体和椎管神经锥体神经元上,介导BZDs的肌肉松弛、中枢性镇静、精神运动性损害以及部分抗惊厥作用。与BZD2(ω2)受体高亲和性的BZDs,具有改善焦虑抑郁症状、镇痛、肌肉松弛、引起共济失调,并可能影响记忆。PBRs被称为BZD3(ω3或Ω3),结构和功能完全不同于中枢BZD受体,分布于全身胶质细胞和大脑干细胞线粒体膜外侧,可能与BZDs的耐受和撤药反应的发生有关,可能参与了一些精神疾病发病过程中神经甾体类因子合成以及线粒体膜渗透性的调节等,在保护神经元免受氧化应激伤害的机制中发挥重要作用[6]。目前是帕金森病、阿尔茨海默病、精神分裂症阴性症状发生机制的重要研究方向之一。
2.2 药代动力学
各种BZDs的不同临床效应除了受到BZDs对GABAA受体α亚单位的选择性结合影响外,还因为不同BZDs的脂溶性和药代动力学特点的差异[1]。
2.2.1 吸收和分布
口服BZDs后,绝大多数BZDs能够从肠道快速和完全(除氯氮䓬外)吸收,生物利用度在80%-99%之间,达峰时间从数十分钟到2小时不等,氯氮䓬在胃内被代谢为活性代谢产物后吸收。
所有的BZDs都是脂溶性药物,可广泛分布于体内脂肪组织中,但不同药物的脂溶性程度不同。影响着药物的肠道吸收以及蛋白结合率。脂溶性较高的BZDs,在肠道的吸收以及进入脑内更迅速,起效也更快。多数BZDs肌内注射的吸收比口服吸收慢,但劳拉西泮和咪达唑仑除外。静脉注射咪达唑仑,可以立即起效。但这种快速吸收和达峰,可能会引起呼吸抑制、低血压、或者疼痛及血栓性静脉炎等,应当谨慎使用。
脂溶性还明显影响BZDs的作用持续时间,因为药物起作用不仅取决于药物进入脑内的快慢和浓度,也取决于脑内和其它部位对药物的清除速率。低脂溶性药物从脑内清除缓慢,因而作用持续时间较长。高脂溶性的药物,如地西泮和阿普唑仑,可以快速从胃肠道吸收,并通过浓度梯度被动扩散快速分布进入脑内快速起效。但是随着脑内药物浓度升高和血中药物浓度的降低,血脑之间的药物浓度梯度出现了逆差,即脑内浓度高于血浆浓度,药物又可快速从脑内排出,药物在脑内的作用随即终止。如果药物的消除半衰期较长,如地西泮,那么尽管药物从脑内排出,药物在脑内的浓度低于其产生效应的水平,但药物仍可在机体内继续留存一段时间。相反,脂溶性低的BZDs,如劳拉西泮,胃肠道的吸收较慢,所以起效也相对慢,但在中枢保持的时间也较长。连续服药达稳态后,脂溶性的影响就会减小甚至消失。
2.2.2 代谢和清除
几乎所有BZDs主要通过肝脏代谢,经过多次生物转化,形成一种或多种活性代谢产物,也有一些药物不经代谢即直接排泄(见图1)。多数BZDs的生物转化方式为氧化(也称为I相代谢),如地西泮、氯氮䓬和氯䓬酸盐等;少数的生物转化方式为与葡萄糖醛酸结合(II相代谢),形成无活性的葡萄糖醛酸甙,或其它硫化或乙酰化物质。有些药物(如地西泮)既有I相代谢也有II相代谢。参与BZDs代谢的细胞色素(CYP)450酶包括3A4、3A5、2B6、2C9和2C19。肝脏疾病、年龄和严重躯体疾病,以及影响氧化功能的一些药物可以影响BZDs的氧化代谢途径。因此对于有明显肝功能损害者、老年人及吸烟者,可以选择(至少在理论上)经II相代谢的药物,因为II相代谢只经过较简单的生物转化形成无活性的代谢产物。奥沙西泮和劳拉西泮比较特殊,是通过结合作用代谢,在老年人和肝脏疾病患者中使用比其他通过氧化作用代谢的BZDs更安全。BZDs对自身代谢没有诱导作用。
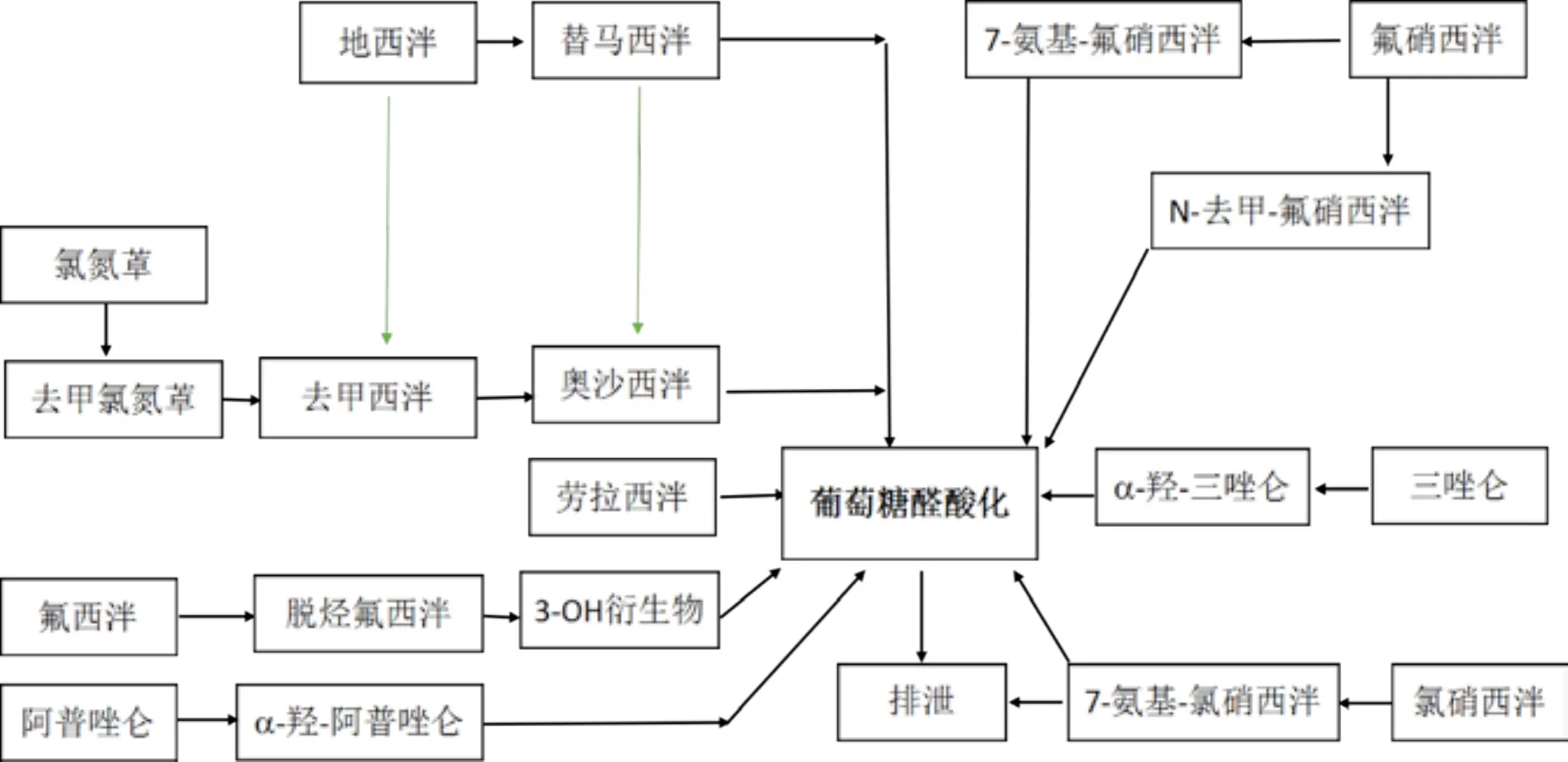
图1 部分BZDs代谢示意图
不同BZDs及其活性代谢产物的半衰期和清除率不同。按照清除半衰期,可将其分为短效、中效及长效三大类。短效BZDs包括半衰期在10小时以内的药物(如三唑仑、咪达唑仑),起效快、作用时间短;中效BZDs的半衰期一般在10-24h之间(如阿普唑仑、奥沙西泮、艾司唑仑),起效速度和作用持续时间介于短效与长效药物之间;而长效药物的半衰期一般在24小时以上,在体内的代谢较慢、作用时间较长(如氯硝西泮、地西泮)。
BZDs的脂溶性、效价和清除半衰期决定了药物在临床应用中的起效时间、疗效持续时间以及给药频率。比较药物的相关特性后,汇总出常用BZDs及非BZDs之间换算的等效剂量,供参考(见表2)。

表2 常见BZDs及非BZDs口服等效剂量表
2.3 使用相关问题
2.3.1 不良反应
BZDs最常见的不良反应主要是困倦、镇静、肌力减低和共济失调。这些不良反应为中枢神经系统受到抑制所致,继续治疗后可逐渐耐受而减轻。较少见的不良反应包括:眩晕,头痛,意识错乱,抑郁,言语不清或构音障碍,性欲改变,震颤,视觉紊乱,尿潴留或尿失禁,胃肠功能紊乱,唾液分泌改变以及失忆等。某些患者可能体验到一种短暂性的兴奋作用,这可使患者出现敌对状态、攻击行为和行为脱抑制。高剂量非胃肠道给药,偶可发生呼吸抑制和低血压。
2.3.2 宿醉效应
指服用长效BZDs后日间出现的镇静与操作能力受损的表现。在临床实践中观察到的现象有时和药理作用不一致,如服用短效药物如三唑仑常见到顺行性遗忘表现,在应对时差所致睡眠紊乱而服用BZDs的患者中,发生的遗忘常被称为“旅行者遗忘”。
2.3.3 过量及处理
BZDs过量中毒后可很快出现意识损害,较为常见的为睡眠样状态,这种情况下患者可被适当刺激短暂唤醒。呼吸抑制极少见或不出现,在不出现缺氧或严重低血压的情况下心率与心律维持正常。由于BZDs脂溶性较高,药物可快速从脑内排出到血浆中,所以即使血药浓度还较高,但患者常可恢复意识。药物急性过量后的恢复期内,患者可出现明显焦虑与失眠。长期用药的患者可能出现伴严重癫痫大发作的戒断综合征。
BZDs过量的处理主要是对症和支持性治疗。只要患者无明显的思睡,可在成人摄入100 mg(或等效剂量)以上、儿童摄入1 mg/kg以上地西泮之后1小时内可给予口服活性碳治疗。单独BZDs过量时不提倡洗胃治疗。对长期使用BZDs或合用三环类抗抑郁药物出现过量的患者,氟马西尼可诱发癫痫发作以及心律失常,此时,一般不主张使用氟马西尼。
2.3.4 依赖与戒断综合征
临床上,即使是短期使用治疗剂量的BZDs,也可能产生药物依赖,尤其是有酒精或药物滥用史和具有明显人格障碍的患者。因此建议用药数周后,应逐渐减少药物剂量,直至停药。使用治疗剂量的BZDs很少出现觅药行为,可能会出现欣快作用。BZDs依赖的危险因素包括:高剂量用药,常规剂量长期用药,使用半衰期短的药物,患者为依赖性人格特质或有药物或酒精依赖史,以及出现药物耐受。突然停用BZDs可能导致严重撤药症状,药物应逐渐减量。对BZDs已经产生依赖的患者,应在专业人员的指导下撤药。
2.3.5 其他使用注意事项
BZDs与其他某些具有中枢神经系统抑制作用的药物合用可能会造成镇静作用的叠加,或增强对呼吸与心血管系统的抑制。多数BZDs经过肝脏CYP450酶代谢,BZDs与那些诱导或抑制CYP450酶的药物合用,有潜在的相互作用风险。此外,已有中枢神经系统抑制或昏迷、呼吸抑制、急性肺动脉瓣关闭不全、重症肌无力、睡眠呼吸暂停及严重肝损害的患者应尽量避免使用BZDs;伴有慢性肺动脉瓣关闭不全、老年患者、伴有肌力减退或肝肾损害的患者应谨慎使用,使用时可能需要降低剂量;地西泮的镇静作用在用药前几天内最为显著,可能会影响到患者驾车或操作机器。地西泮和其他BZDs不建议在青光眼患者中使用,但这一禁忌证的原理尚不明确。
3 临床常用的BZDs
临床使用的BZDs多达50余种,在我国临床实践中,常用的不超过10种,本文主要介绍在我国临床中最常使用的几种BZDs。
3.1 地西泮
地西泮(diazepam)是临床上最常用的抗焦虑药物之一,给药方式有口服、肌内或静脉注射形式。口服吸收迅速而完全,达峰时为30至90分钟。肌内注射给药吸收较不稳定,血浆峰浓度低于口服给药。血浆蛋白结合率为98%-99%。地西泮主要在肝脏经CYP2C19同工酶代谢,包括I相和II相代谢途径,消除半衰期1-2天;其主要活性代谢物去甲西泮的半衰期更长(2至5天),所以地西泮的作用时间可被进一步延长。重复给药后地西泮和去甲西泮在体内可积聚,长期给药后体内去甲西泮的相对比例增加。地西泮主要以游离或代谢物的形式经过肾脏排泄。在新生儿、老年人以及患有肝病的患者,地西泮和/或其代谢产物的血浆清除半衰期延长。因此,过量或长期服用可能会出现较大的毒副反应,主要表现为中毒及依赖性等,严重时会出现撤药综合征。地西泮及其代谢产物也可透过胎盘屏障,并可分布于母乳中。
2013年,《电子招标投标办法》出台,提出了推进电子投标招标的必要性,从而减少政府采购的成本,提高采购资金的使用效率。2016年,《“互联网+”招标采购行动方案(2017~2019年)》出台,提出通过互联网推进电子投标招标的发展,使得招标过程更加智能化、透明化,提高招标效率,从而能推进招标代理行业的发展。
地西泮属于长效、中等强度的BZDS,具有镇定、催眠、抗焦虑、抗惊厥和肌肉松弛作用。卟啉病患者、肝肾损害的患者慎用。单独过量服用地西泮极少导致患者死亡。
3.2 奥沙西泮
奥沙西泮(oxazepam)口服能被胃肠道很好地吸收,但吸收速度较慢,达峰时约2小时。生物利用度99%,血浆蛋白结合率为85~97%,清除半衰期5~12小时。奥沙西泮为多种BZDs如地西泮、普拉西泮、替马西泮的代谢产物。该药不经过CYP450代谢,而是在肝脏与葡萄糖醛酸直接结合成为非活性的葡萄糖苷酸奥沙西泮,代谢不会受年龄和轻/中度肝损害的影响,通过肾脏排出。所有BZDs治疗肝损伤患者时都应谨慎,而奥沙西泮相对安全。肾损伤患者不需要调整奥沙西泮的用量。该药能通过胎盘且可分泌到母乳中。
奥沙西泮是一种中、短半衰期BZDs,具有和地西泮相似的一般药理特性。有镇静、催眠、抗惊厥、抗焦虑、缓解酒精戒断症状等作用。该药没有活性代谢产物,长期给药较少在体内蓄积,相互作用少。
3.3 艾司唑仑
艾司唑仑(estazolam)口服后吸收良好,平均达峰时2小时,血浆蛋白结合率约为93%;平均清除半衰期为10-24小时。艾司唑仑经肝脏CYP3A4广泛代谢,主要代谢产物有无药理活性的4-羟艾司唑仑和1-氧艾司唑仑。这些代谢产物以游离或结合的形式经肾脏排泄,在粪便中也可检测到少量,只有少部分药物以原形排出。
3.4 劳拉西泮
劳拉西泮(lorazepam)口服吸收完全,约2小时达血浆峰浓度,生物利用度约90%。肌内注射与口服使用后的药物吸收曲线相似。血浆蛋白结合率为85%,可透过血脑屏障和胎盘屏障,并能分泌到母乳中。与奥沙西泮类似,劳拉西泮不经过CYP450代谢,而是在肝脏与葡萄糖醛酸直接结合成为非活性的葡萄糖苷酸劳拉西泮,通过肾脏排泄,消除半衰期为11-16小时。
静脉注射劳拉西泮,在不同的媒介(如水,葡萄糖和氯化钠注射液)中溶解度不同,溶解度最大的是5%的葡萄糖注射液,溶解度为62μg/ml;最小的是0.9%的氯化钠注射液,溶解度为27μg/ml。将劳拉西泮注射液用0.9%的氯化钠注射液稀释到浓度为0.5mg/ml时,会出现沉淀。这种溶解度的差异可能和液体的PH值有关,临床使用的劳拉西泮注射液,在丙二醇中加入了聚乙二醇。
劳拉西泮是一种高效能中短效的BZDs,具有和地西泮相似的一般药理特性。该药的抗焦虑作用在BZDs中最强,是地西泮的2-5倍,抗惊厥效果良好,在临床中也经常用来辅助抗精神病药物治疗急性兴奋激越。长期静脉使用劳拉西泮,可能因聚乙二醇或丙二醇诱发一定的不良反应。严重肝损伤的患者,可能明显延长劳拉西泮的半衰期,禁忌使用;轻中度肝损伤的患者,可减量使用。
3.5 咪达唑仑
咪达唑仑(midazolam)吸收迅速,根据给药途径的不同,达峰时间从20分钟到60分钟不等。有明显首过效应,口服剂的生物利用度较低,肌内注射生物利用度约90%但不稳定。血浆蛋白结合率约96%。咪达唑仑在肝脏经CYP3A4同工酶代谢,主要代谢产物α-羟咪达唑仑有一定的活性,半衰期小于1小时。咪达唑仑的代谢产物主要以葡萄糖醛酸苷结合物的形式经肾脏排泄,消除半衰期为2-7小时。
咪达唑仑是一种短效BZDs,一般药理特性和地西泮相似,但引起遗忘的作用更强。已有报道静脉给予咪达唑仑达到抑制知觉效果后,患者由于呼吸抑制、低血压或心跳停止而死亡,因此静脉注射咪达唑仑应小心。尽管使用等效剂量时,咪达唑仑和地西泮引起的氧饱和度下降程度相似,但咪达唑仑引起的抑制效果更突然。因此建议静脉使用咪哒唑仑时应当谨慎,如备好复苏设备、持续监控心肺功能、谨慎加量等,尤其是同时接受麻醉性镇痛药治疗的患者、老年和儿童患者,以及心肺功能差的患者更应小心。口服咪达唑仑,也有类似的注意事项。
3.6 阿普唑仑
阿普唑仑(alprazolam)口服吸收良好,单次服用后血浆浓度在1~2小时达峰值。平均血浆半衰期为11~15小时。血浆蛋白结合率为70%~80%,主要与血浆白蛋白结合。阿普唑仑主要由肝CYP450同工酶CYP3A4代谢,代谢产物α-羟-阿普唑仑的血浆浓度较低,活性相当于母药的一半。阿普唑仑以原始药物和代谢产物的形式经尿液排出。
阿普唑仑(alprazolam)是高效价、中效(中等半衰期)BZDs,通常被用于焦虑障碍患者。停用阿普唑仑时常会引起焦虑症状的反跳。在脑外体循环中,阿普唑仑具有致卟啉原的作用,卟啉病患者慎用,肝肾损害的患者慎用。
3.7 氯硝西泮
氯硝西泮(clonazepam)口服容易吸收,1-4小时可达血浆峰值,生物利用度约90%,血浆蛋白结合率约85%。氯硝西泮在肝脏中被完全代谢,主要代谢产物无抗惊厥作用。氯硝西泮以代谢产物的游离型或结合型几乎完全从肾脏排泄。消除半衰期为20-40小时或更长。氯硝西泮可透过胎盘屏障,并可分布于乳汁中。和其它抗精神病药合用,可能会影响氯硝西泮的体内代谢过程。在健康受试者中发现肌内注射比口服吸收慢,且个体间差异较大。
氯硝西泮(clonazepam)是一种BZDs衍生物,具有镇静、催眠、抗焦虑和抗惊厥等作用,其一般药理作用类似于地西泮,抗惊厥作用比硝西泮强5倍。国内外药典将此药收录在抗惊厥药中。该药可用于各种类型癫痫发作的治疗,由于其潜在的耐受性和镇静作用,长期治疗常选用其它抗惊厥药。也用于治疗肌阵挛及相关的异常运动以及惊恐障碍。
氯硝西泮的不良反应与地西泮相似,但是困倦更常见,唾液或支气管分泌过多可能导致儿童呼吸道问题。静脉给药有发生血栓性静脉炎的风险,通常建议选择大静脉、缓慢给药(给药速度不超过0.5mg/分钟),并且监测呼吸和血压。停用时需监测患者反应,逐渐减停,以降低撤药反应。
4 小结
BZDs是一类核心结构和作用靶点相似的常用药物,在临床上用于治疗焦虑、失眠、癫痫发作、肌肉痉挛以及麻醉前辅助用药等。但是不同BZDs的理化性质、药代动力学特征以及中枢药理机制仍然存在差异,了解常用BZDs的药理学特征,有助于临床合理应用BZDs。
[1] Sadock BJ, Sadock VA, ruiz P. Kaplan&Sadock's Comprehensive textbook of psychiatry. 9th Edition. Philadelphia: Lippincott Williams &Wilkins, 2009.
[2] Kaufmann WA, Humpel C, AlheidGF, et al. Compartmentation of alpha 1 and alpha 2 GABAA receptorsubunits within rat extended amygdala: implications for benzodiazepine action. Brain Res, 2003, 964(1):91-99.
[3] John R Atach. The benzodiazepine binding site of GABAA receptors as a target for the development of novel anxiolytics. Expert Opinion on Investigational Drugs, 2005, 14(5): 601-618.
[4] McKernan RM., Rosahl TW, Reynolds DS, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABAA receptor α1 subtype. Nature Neuroscience, 2000, 3: 587 – 592.
[5] Gabriella Guerrini, Giovanna Ciciani. Benzodiazepine receptor ligands: a patent review (2006-2012). Expert Opin. Ther patents, 2013, 23(7): 843-866.
[6] HannsMohler. The Legacy of the Benzodiazepine Receptor: from flumazenil to enhancing cognition in Down syndrom and social interaction in autism. Advances in pharmacology, 2015, 72: 1-36.
10.15900/j.cnki.zylf1995.2017.02.003
2017-01-03)
司天梅(1968.06-),女,研究生学历,精神科主任医师,教授,北京大学第六医院研究室主任。主要研究方向:精神药理学和临床治疗学。E-mail:si.tian-mei@163.com.
