LC-MS/MS法同时测定人血浆中咪达唑仑和1-羟基咪达唑仑的浓度*
2021-09-01黄烽如房秋晨孙鲁宁王永庆
沈 叶,黄烽如,房秋晨,孙鲁宁**,王永庆**
1 徐州医科大学 江苏省新药研究与临床药学重点实验室,徐州 221004;2 南京医科大学第一附属医院 临床药理研究室,南京210029
细胞色素P450(cytochrome P450,CYP450)是人体最重要的药物代谢酶之一,其主要亚型(CYP3A4/5)参与50%以上临床用药的代谢[1,2]。许多外源性物质可以作为诱导剂或抑制剂影响CYP3A底物的体内暴露。因此,确定CYP3A 及其同工酶的活性对于评估药物相互作用至关重要[3]。目前,评价人体内CYP3A 酶活性的方法主要有探针药物法[4](如咪达唑仑、氨苯砜、红霉素等)和内源性生物标志物法[5,6]。咪达唑仑被我国药监局和美国药品监督管理局药品审评中心推荐为CYP3A 酶活性评价的首选探针底物[7]。咪达唑仑(1-甲基-8-氯-6-(2-氟苯基)-4H-咪唑并[l,5-a][l,4]苯并二氮杂艹卓)作为使用最为广泛的苯二氮唑类短效中枢神经系统抑制剂,具有镇静、催眠、抗惊厥、肌肉松弛的作用[8]。咪达唑仑作为探针药物进入人体后经CYP3A 代谢生成主要代谢物1-羟基咪达唑仑[9]。
目前,文献报道测定人血浆中咪达唑仑的分析方法主要为高效液相色谱法[10,11]和液相色谱-串联质谱法[8,12~14]。然而,文献[10,11]中建立的方法灵敏度较低,分析时间较长。文献[13,14]开发的方法前处理复杂,需要萃取或者旋干的操作,分析时间较长。现建立了一种前处理简单快捷、线性范围宽、高效灵敏的同时测定人血浆中咪达唑仑和1-羟基咪达唑仑的HPLC-MS/MS 法,可应用于咪达唑仑作为CYP3A 探针的临床药物相互作用的研究。
1 仪器与药品、试剂
1.1 仪器
1290 Infinity 高效液相色谱仪(美国安捷伦科技有限公司),包括G4220A 二元高压泵;G1316C 柱温箱和G4226A 多孔板自动进样器;API 4000 三重四极杆质谱仪,配备电喷雾离子源,Analyst1.6.3 数据采集处理系统(美国应用生物公司);BP211D 电子天平(德国Sartorius 公司);Mixmate 涡旋混匀仪(德国Eppendorf 公司);Stratos 离心机(德国Thermo Fisher 公司)。
1.2 药品与试剂
马来酸咪达唑仑(中国食品药品检定研究院,批号:E1822047,纯度:100.0%);1-羟基咪达唑仑溶液(美国Cerilliant 公司,批号:FN12081503,浓度:100 μg·mL-1,溶剂:甲醇);d4-咪达唑仑溶液(美国Cerilliant 公司,批号:FE03311701,浓度:100 μg·mL-1,溶剂:甲醇);乙腈为色谱纯(美国Merck 公司);超纯水(18.2 MΩ)由Millipore 纯水仪制得。
2 方法与结果
2.1 色谱条件
色谱柱:Kinetex C18(2.1mm×50mm,2.6μm);柱温:38℃;流动相由1mmol·L-1甲酸铵溶液(含0.1%甲酸)(A 相)和乙腈(B 相)组成,梯度洗脱程序见表1;流速:0.3mL·min-1;进样量:3.0μL;分析时间7min。
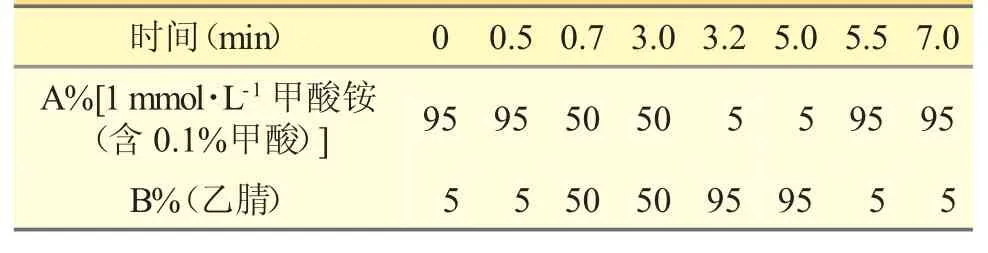
表1 梯度洗脱程序
2.2 质谱条件
采用多重反应监测(MRM)离子检测方式,离子化方式:电喷雾离子化(ESI),在正离子模式下进行扫描检测,离子化电压为4500 V,温度设为600 ℃,雾化气压力为60 psi,涡轮气压力为60 psi,气帘气压力为50 psi,碰撞气压力为10 psi。咪达唑仑、1-羟基咪达唑仑及d4-咪达唑仑的定量离子对,去簇电压(DP),射入电压(EP),碰撞能量(CE),碰撞室射出电压(CXP)见表2。

表2 质谱条件
2.3 储备液和工作液的配制
精密称取马来酸咪达唑仑对照品13.55 mg(经纯度换算相当于咪达唑仑10.00 mg),用甲醇溶解定容至10.0 mL 量瓶中,得到1.00 mg·mL-1的咪达唑仑储备液。取咪达唑仑储备液40 μL 以及100 μg·mL-1的1-羟基咪达唑仑储备液200 μL 置于同一2.0 mL 量瓶中,并用甲醇定容,得到咪达唑仑和1-羟基咪达唑仑浓度分别为20.0/10.0 μg·mL-1的混合工作液,将此混合工作液用甲醇逐级稀释,得到咪达唑仑和1-羟基咪达唑仑浓度分别为6.00/3.00、8.00/4.00、80.0/40.0、400/200、1200/600、2000/1000、3200/1600、4000/2000 ng·mL-1的系列浓度标准曲线用混合工作液,以及浓度分别为16.0/8.00、1000/500、3600/1800 ng·mL-1的质控用混合工作液。
以乙腈为溶剂,配制含d4-咪达唑仑浓度为15.0 ng·mL-1的内标工作液。
2.4 标准曲线和质控样本的配制
采用空白血浆配制标准曲线和质控样本。取50 μL 空白血浆,加入2.50 μL 相应浓度的混合工作液,配成含咪达唑仑和1-羟基咪达唑仑浓度分别为0.300/0.150、0.400/0.200、4.00/2.00、20.0/10.0、60.0/30.0、100/50.0、160/80.0、200/100 ng·mL-1的混合标准曲线样本,以及咪达唑仑和1-羟基咪达唑仑浓度分别为0.800/0.400 ng·mL-1、50.0/25.0 ng·mL-1和180/90.0 ng·mL-1的混合质控样本。
2.5 样本前处理
取50 μL 血浆样本,加入150 μL 含d4-咪达唑仑的乙腈工作液,置1.5 mL 离心管中,涡旋10 min,16000 r·min-1离心15 min(4 ℃),取上清液进样作LC-MS/MS 分析。
2.6 专属性考察
本实验分别考察了咪达唑仑和1-羟基咪达唑仑及d4-咪达唑仑在人血浆中的专属性。咪达唑仑和1-羟基咪达唑仑及d4-咪达唑仑的保留时间分别约为2.3 min、1.9 min 和2.3 min。典型色谱图见图1。结果表明,血浆中咪达唑仑和1-羟基咪达唑仑及d4-咪达唑仑峰位处无干扰。
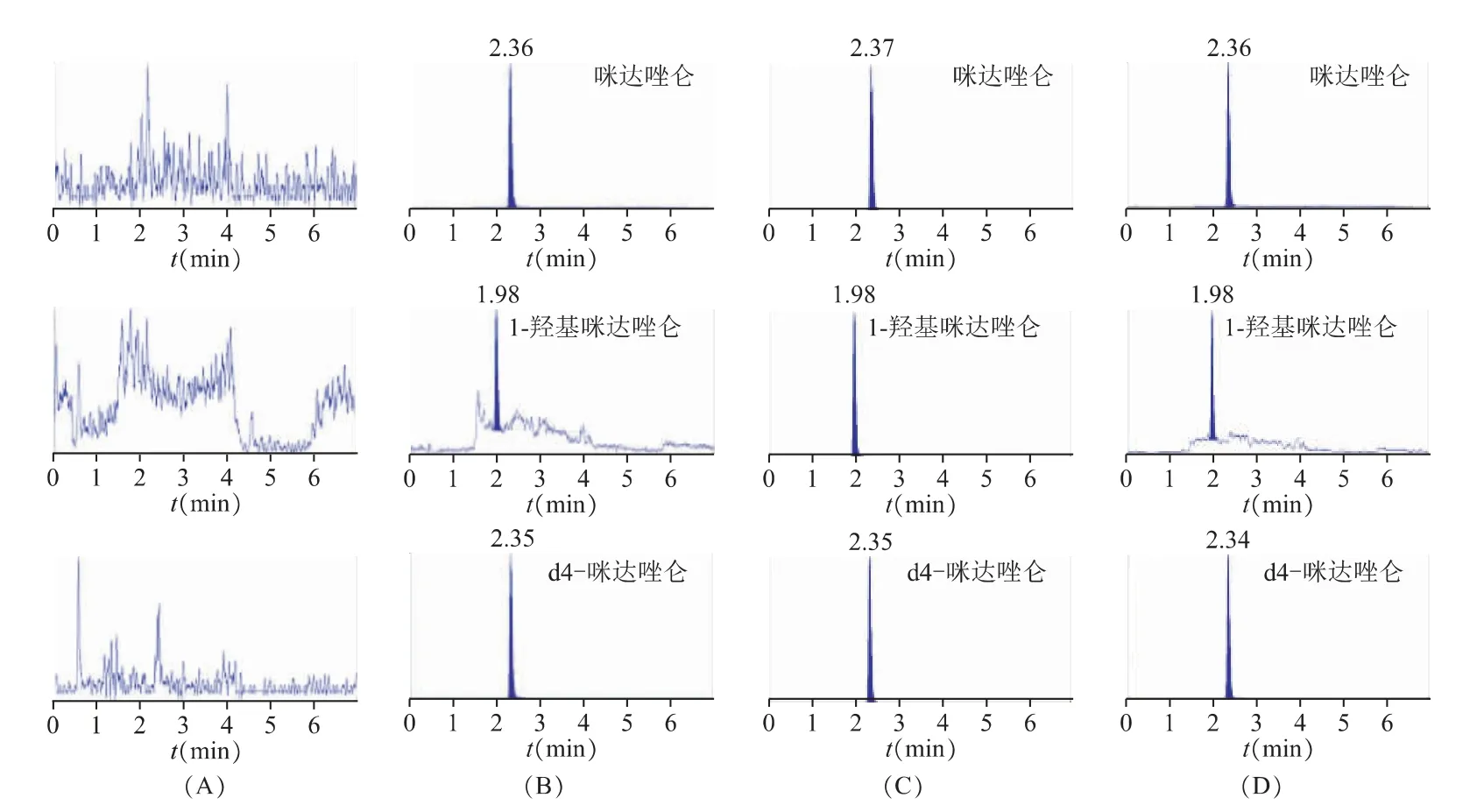
图1 咪达唑仑和1-羟基咪达唑仑及d4-咪达唑仑在血浆中的色谱图
2.7 标准曲线
按“2.4”项方法制备混合标准曲线样本和混合质控样本。按“2.5”项下方法处理样本。以浓度(C)为横坐标,以待测化合物/内标峰面积比值(f)为纵坐标,用权重系数1/C2进行最小二乘法作线性回归。典型的咪达唑仑和1-羟基咪达唑仑标准曲线方程分别为:

结果表明,分别在0.300~200 ng·mL-1与0.150~100 ng·mL-1线性关系良好。
2.8 提取回收率
按“2.4”项下方法,使用6 个不同来源的空白血浆制备低浓度(咪达唑仑/1-羟基咪达唑仑,0.800/0.400 ng·mL-1)、中浓度(咪达唑仑/1-羟基咪达唑仑,50.0/25.0 ng·mL-1)、高浓度(咪达唑仑/1-羟基咪达唑仑,180/90.0 ng·mL-1)混合质控样本,按“2.5”项下方法处理。同时在6 个不同来源的空白血浆的乙腈提取液加入相同浓度的咪达唑仑、1-羟基咪达唑仑和d4-咪达唑仑工作液,涡旋混匀。上述每个浓度样本各配制3 份,进样分析后,以空白血浆制备的样本中分析物的峰面积与空白血浆的乙腈提取液制备的样本中分析物的峰面积的比值,评价提取回收率。结果显示,3 种浓度血浆样本中咪达唑仑的提取回收率分别为(101.6±6.6)%、(102.2±2.9)%、(100.6±6.8)%;1-羟基咪达唑仑的提取回收率为(101.8±4.6)%、(102.4±3.2)%、(101.1±7.1)%。
2.9 基质效应
按“2.4”和“2.5”项下方法,使用6 个不同来源的空白血浆制备乙腈提取液,加入待测咪达唑仑、1-羟基咪达唑仑和d4-咪达唑仑工作液,制备低浓度(咪达唑仑/1-羟基咪达唑仑,0.800/0.400 ng·mL-1)和高浓度(咪达唑仑/1-羟基咪达唑仑,180/90.0 ng·mL-1)质控的基质效应样本;同时用水基质制备相同的低浓度和高浓度样本;将上述样本进样分析,得到各样本待测化合物与内标的峰面积,计算待测化合物/内标峰面积比值(f),用于评价血浆基质对分析的影响。
在低浓度和高浓度血浆样本中,咪达唑仑的基质效应分别是(101.4±4.3)%、(98.9±2.7)%,1-羟基咪达唑仑的基质效应分别是(99.1±3.0)%、(95.5±2.9)%,结果显示,血浆基质不影响样本检测。
2.10 准确度和精密度试验
按“2.4”项下方法,配制定量下限样本,低、中、高3 种浓度的血浆基质混合质控样本,每个浓度配制5 份(n=5),按“2.5”项下方法处理,考察批内和批间准确度和精密度。结果见表3。

表3 精密度和准确度试验结果(n=5)
2.11 稳定性试验
按“2.4”项下方法,配制浓度为低、高两种浓度的血浆基质混合质控样本,分别在室温(24 ℃)放置12 h、自动进样器(10 ℃)中放置24 h、3 次冻融循环、-40 ℃冰冻10 个月,按“2.5”项下方法处理,以准确度评估稳定性。结果显示,待测化合物在上述各条件下稳定性均良好。见表4。

表4 稳定性试验结果
3 人血浆中咪达唑仑和1-羟基咪达唑仑浓度的测定
评价促变药物对马来酸咪达唑仑片在健康受试者中的药代动力学影响研究(预试验),在南京医科大学第一附属医院Ⅰ期临床试验室完成。在第一周期研究中,6 例健康男性受试者于试验第1 天早上空腹口服15 mg 马来酸咪达唑仑片(1 片),分别于给药前0 h(给药前1.0 h 内)和给药后10、20、30、45 min,1.0、1.5、2.0、2.5、3.0、4.0、5.0、8.0、12、14、24 h(共16 个时间点),采集上肢静脉血4 mL,分离血浆后保存于-80°C 冰箱待用。使用建立的LC-MS/MS 法对上述血样进行浓度测定。6 例健康男性受试者咪达唑仑及1-羟基咪达唑仑血药浓度均值-时间曲线见图2。
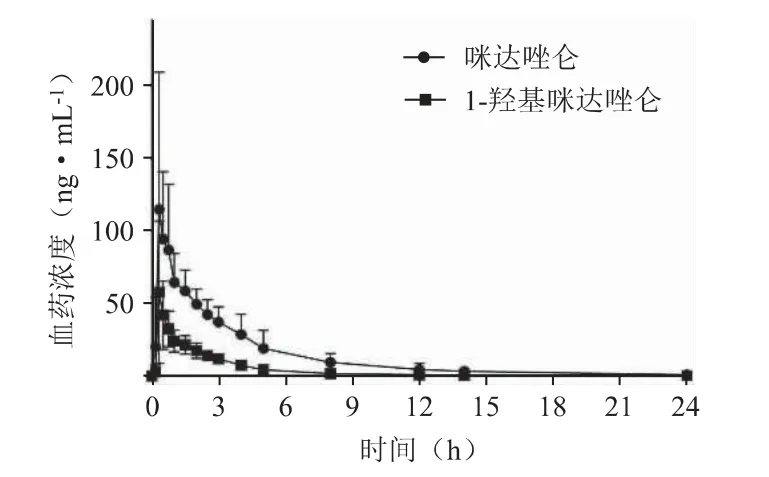
图2 健康男性受试者咪达唑仑及1-羟基咪达唑仑血药浓度均值-时间曲线(n=6)
4 讨论
本研究建立并验证了同时测定人血浆中咪达唑仑和1-羟基咪达唑仑的HPLC-MS/MS 法。在方法开发过程中,对每项质谱参数进行了优化,使得两个待测化合物均具现有条件下最高的质谱响应。
在优化色谱条件时,考察了用乙腈和甲醇作为有机相的方法,发现两种有机试剂对于峰型和响应的影响相差无几;但考虑到乙腈沉淀蛋白效果更佳,因此在有机相的选择上最终确定为乙腈。除此之外还评价了1 mmol·L-1乙酸铵和1 mmol·L-1乙酸铵(含0.1%甲酸)作为水相的方法,研究发现,甲酸的加入会大大减轻残留效应,因此最终选择乙腈-1 mmol·L-1甲酸铵(含0.1%甲酸)作为流动相。流速设置为0.3 mL·min-1可以同时兼顾分析时间和色谱柱压力,并且具有良好的分离效果。同时建立了梯度洗脱程序,在实现良好的色谱分离的同时,在待测化合物出峰后使用95%有机相冲洗1.8 min 以保护色谱柱和减少本次进样对下一次进样的影响,以提高方法的耐用性。
本方法仅需50 μL 的样本量,样品制备程序基于一个简单的蛋白质沉淀步骤,避免了复杂的萃取过程,灵敏度较高,且分析时间较短。最终建立了同时测定人血浆中咪达唑仑和1-羟基咪达唑仑浓度的HPLC-MS/MS 法,可用临床药物相互作用研究。
