维罗非尼联合爱必妥对结直肠癌细胞株的作用研究
2017-06-01曲秀娟车晓芳郭天舒刘云鹏
马 瑞, 徐 玲, 曲秀娟, 车晓芳, 郭天舒, 刘云鹏
(中国医科大学 附属第一医院,辽宁 沈阳 110001)
维罗非尼联合爱必妥对结直肠癌细胞株的作用研究
马 瑞, 徐 玲, 曲秀娟, 车晓芳, 郭天舒, 刘云鹏*
(中国医科大学 附属第一医院,辽宁 沈阳 110001)
探讨BRAF抑制剂维罗非尼联合EGFR单克隆抗体西妥昔单抗对BRAF突变型和野生型结直肠癌细胞株的作用。应用MTT法检测,将不同浓度西妥昔单抗、维罗非尼单药以及两药联合分别作用于BRAF突变型结直肠癌细胞株RKO、HT-29和BRAF野生型结直肠癌细胞株DIFI、CACO-2后,观察各细胞株增殖变化;通过集落形成实验,经过长时间培养细胞验证不同处理因素对细胞增殖的抑制作用;Western blot实验检测EGFR下游通路靶蛋白的活性在两药联合后是否出现明显下调。结果显示,单药治疗,西妥昔单抗能一定程度抑制BRAF野生型结直肠癌细胞株增殖,抑制率达26%~40%,而BRAF突变组两种细胞株对西妥昔单抗均耐药。维罗非尼对不同基因状态细胞的增殖均有一定的抑制作用,IC50均小于7 μmol/mL,其中BRAF突变细胞更为敏感。对比单药西妥昔单抗,联合维罗非尼后更加明显地抑制了细胞增殖,4种细胞存活率均明显降低(P<0.05)。长时间集落形成实验证实两药联合形成集落的面积和数量均明显减少。Western Blot实验显示对比单药,两药联合明显下调了p-EGFR、BRAF、p-ERK的表达。结果显示,BRAF抑制剂维罗非尼无论在BRAF突变型还是野生型结直肠癌细胞株中,均能增强西妥昔单抗作用的敏感性。
西妥昔单抗;维罗非尼;EGFR;BRAF;结直肠癌
结直肠癌(Colorectal cancer,CRC)是世界范围内最常见的消化系统恶性肿瘤之一,统计显示,目前结直肠癌的发病率和死亡率均居各种常见恶性肿瘤前列。研究表明,CRC在发生、发展过程中与多条信号转导通路的活化密切相关,其中表皮生长因子受体(Epidermal growth factor receptor,EGFR)介导的通路尤为重要。EGFR是一种跨膜酪氨酸激酶受体,广泛分布于人体各种组织器官的细胞中。与配体结合可引起其异常激活,从而引起下游信号转导通路的活化,进而诱导肿瘤细胞增殖、侵袭、转移以及血管生成[1]。EGRR单克隆抗体,西妥昔单抗(爱必妥,Cetuximab)通过作用于EGFR胞外区抗体位点干扰二聚体形成,进而诱导受体内化及下调。其主要功能是阻断EGFR信号转导,减低酪氨酸激酶的活性,从而抑制肿瘤的存活、增殖、侵袭和转移[2]。爱必妥还可以发挥免疫抗肿瘤的作用,其激发免疫效应细胞对肿瘤靶细胞的杀伤作用,通过介导抗体依赖细胞介导的细胞毒性作用(Antibody dependent cell-mediated cytotoxicity,ADCC)靶向杀伤表达 EGFR 的肿瘤细胞[3]。 ADCC 作用指表达IgG Fc受体的靶细胞膜抗原与已结合在肿瘤细胞表面的特异性 IgG 类抗体结合后,通过抗体Fc段构象的改变与效应细胞表面的FcγR结合,活化效应细胞,进而触发其对靶细胞的杀伤作用。但爱必妥单药治疗转移性结直肠癌(mCRC)的有效率十分有限,仅为8%~13%[4],即使与mCRC的主要化疗方案FOLFOX、FOLFIRI联合用药,有效率较常规化疗方案也仅提高8%~16%[5]。导致爱必妥敏感性不高的重要原因是耐药发生。RAS基因突变(KRAS、NRAS、HRAS)是目前公认的原发性耐药机制。而有些RAS野生型患者仍对爱必妥耐药,对RAS的下游信号进行探索发现,约有10%的mCRC患者伴有鼠类肉瘤滤过性毒菌致癌同源体B1基因(v-Rafmurine sarcoma viral oncogene homolog B1,BRAF)突变。BRAF蛋白激酶是有丝分裂原活化蛋白激酶(MAPK)通路上重要的信号转导因子,其涉及到多种肿瘤发生发展及侵袭转移过程[6]。现已于多种恶性肿瘤中发现了BRAF基因的突变,包括恶性黑色素瘤、甲状腺癌、结直肠癌、肺癌、毛细胞白血病等。研究发现BRAF基因突变发生率最高的是V600E 的突变,有90%以上肿瘤为此类突变。目前尚未开展过评价BRAF突变状态与抗EGFR治疗关系的随机临床试验,然而越来越多临床研究的亚组分析显示,BRAF突变的mCRC患者比BRAF野生型者对爱必妥的反应更低,无疾病进展生存期 (PFS) 和总生存期 (OS) 更短,疾病控制率也不理想[7]。近期研制的小分子TKI类靶向药物维罗非尼,特异性地与BRAF V600E相结合,通过封闭下游通路抵抗肿瘤细胞的增殖,其在EGFR低表达的黑色素瘤的疗效较好。目前维罗非尼已被FDA批准治疗晚期恶性黑色素瘤患者,然而其在同样含有BRAF突变的结直肠癌治疗中收效甚微,反应率仅为5%左右[8]。目前认为维罗非尼作用于EGFR表达水平较高的CRC细胞则会反馈性激活EGFR进而激活下游的RAS及CRAF,而对其产生原发耐药。目前无论临床实验还是基础研究均显示,对BRAF突变的mCRC患者应用爱必妥或帕尼单抗联合维罗非尼治疗可提高疗效[9-10]。而维罗非尼在BRAF野生型患者的治疗中扮演怎样的角色,是否可以通过与爱必妥联合作用加强抗肿瘤效果尚无明确报道。为了探讨维罗非尼是否能够通过联合用药提高爱必妥对结直肠癌治疗的敏感性及其机制,本研究将单药爱必妥、单药维罗非尼及两药联合作用于结直肠癌BRAF突变及野生型细胞株中,探讨联合用药效果、作用机制,以及对EGFR/RAF/RAS/ERK通路的影响。
1 材料与方法
1.1 材料
人结直肠癌细胞株RKO、HT-29为BRAFV600E突变型、KRAS野生型细胞,人结直肠癌细胞株DIFI、CACO-2为BRAF、KRAS野生型细胞,以上细胞株均购自中国科学研究院。爱必妥购自EMD Millipore公司(Billerica, MA, USA),维罗非尼购自Selleck公司,兔抗人p-EGFR、p-ERK、BRAF抗体购自Cell Signaling公司,兔抗人EGFR、ERK 和β-actin 抗体购自 Santa Cruz 公司,RPMI 1640培养基购自Gibco公司。胎牛血清购自中国医学科学院血液学研究所。
1.2 方法
1.2.1 细胞培养 RKO、HT-29、DIFI和CACO-2为常规传代培养,用含有10%胎牛血清、青霉素(100 U/mL)和链霉素(100 U/mL)的RPMI-1640,于37 ℃、5%CO2、饱和湿度的培养箱内培养。2~3 d传代1次。
1.2.2 MTT法检测细胞活力 将细胞接种于96孔板,细胞贴壁后换液,分别加入一定浓度的爱必妥和/或维罗非尼,每组设3个复孔,每孔终体积为200 μL。分别培养48 h和72 h后每孔加入MTT 溶液20 μL,孵育4 h后弃上清,每孔加入200 μL DMSO,振荡摇匀。最后测定吸光度并计算细胞活力。
1.2.3 集落形成实验 将增殖活跃特定数量的4种结直肠癌细胞接种于12孔板上(500个/孔),细胞培养24 h后先加入维罗非尼(2 μmol/mL),1 h后加入爱必妥(10 μg/mL)。细胞培养14 d后弃上清,采用瑞氏-吉姆萨法进行染色,并采用象限计数的方法计算细胞集落数量。
1.2.4 Western blot 检测蛋白变化 收集细胞,PBS洗2次后加入1% Triton裂解液,冰上裂解5 min,高速离心(13 000 r/min) 25 min,弃沉淀,进行蛋白定量。在样品中加入3×buffer并相互混合,放入煮样器煮沸5 min,进行蛋白电泳和转印,之后牛奶封闭1 h。加入p-EGFR、EGFR、BRAF、ERK、p-ERK及β-actin的一抗,4 ℃孵育过夜。1×TTBS洗10 min×4次后,二抗(1∶ 2 000)室温封闭30 min,1×TTBS再洗10 min×4次后,采用ECL法显色,并分析处理。
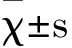
2 结果与分析
2.1 爱必妥对结直肠癌细胞株增殖的影响
0、0.1、1.0、10.0、100.0 μg/mL爱必妥分别作用于BRAF突变型和野生型结直肠癌细胞 48和72 h 后,应用 MTT 方法检测爱必妥对细胞增殖的影响。结果显示爱必妥作用于BRAF突变型细胞几乎不能引起细胞死亡,细胞的存活率均不低于80%(图1)。提示BRAF突变结直肠癌细胞株对爱必妥耐药。而爱必妥作用于BRAF野生型结直肠癌细胞株,细胞增殖抑制率达26%~40%,提示BRAF野生型结直肠癌细胞株对爱必妥作用敏感(图2)。
2.2 维罗非尼对结直肠癌细胞株增殖的影响
0、0.1、1.0、10.0、50.0 μmol/mL 维罗非尼分别作用于BRAF突变型和野生型结直肠癌细胞48和72 h 后,应用MTT 方法检测维罗非尼对细胞增殖的影响。结果显示维罗非尼无论对BRAF突变型还是野生型结直肠癌细胞均有增殖抑制作用,而BRAF突变细胞更加显著(图3、图4)。各细胞株IC50结果见表1。提示无论BRAF突变还是野生型结直肠癌细胞株对维罗非尼均较敏感。

图1 爱必妥对BRAF突变细胞株抑制率Fig.1 Inhibition rate of cetuximab on BRAF mutant cell lines

图2 爱必妥对BRAF野生细胞株抑制率Fig.2 Inhibition rate of cetuximab on BRAF wild cell lines

图3 维罗非尼对BRAF突变细胞株抑制率Fig.3 Inhibition rate of vemurafenib on BRAF mutant cell lines

表1 各个细胞株基因突变情况及维罗非尼作用细胞的IC50

图4 维罗非尼对BRAF野生细胞株抑制率Fig.4 Inhibition rate of vemurafenib on BRAF wild cell lines
2.3 维罗非尼增强爱必妥对结直肠癌细胞的敏感性
维罗非尼(2 μmol/mL)联合爱必妥(10 μg/mL)作用72 h 后,与爱必妥单药组相比,细胞存活率RKO从(93.71±3.61)% 降至(38.51±2.31)%,HT-29 从(86.22±4.86)% 降至(28.11± 3.32)%,DIFI从(77.21±2.44)%降至(28.45±6.12)%,CACO-2 从(82.55±4.64)%降至(20.57±3.57)%。联合治疗组与单药治疗组相比均有明显的统计学差异,P<0.05 (图5、图6)。在长时间集落形成实验中,两药联合也能明显抑制细胞集落的形成(图7)。维罗非尼(2 μmol/mL)联合爱必妥(10 μg/mL)作用24 h,p-EGFR、BRAF、p-ERK 均较单药作用出现明显下调(图8)。结果表明,维罗非尼通过联合作用抑制EGFR及其通路提高了BRAF突变及野生型结直肠癌细胞对爱必妥作用的敏感性。

图5 两药联合对BRAF突变细胞株抑制率Fig.5 Inhibition rate of combination on BRAF mutant cell lines*P<0.05,下图同

图6 两药联合对BRAF野生细胞株抑制率Fig.6 Inhibition rate of combination on BRAF wild cell lines

图7 4个细胞系集落形成实验Fig.7 Colony-forming assay of 4 cell lines

图8 两药联合明显下调EGFR及其下游通路Fig.8 Two drugs significantly lowered EGFR and downstream pathway
3 讨 论
随着生活水平不断提高,结直肠癌患者数量在世界范围内不断攀升,目前结直肠癌仍是威胁人类生命的健康杀手。爱必妥作为首个获批用于进展期结直肠癌治疗的抗EGFR单克隆抗体,通过竞争性并特异性地结合肿瘤细胞表面的EGFR受体,有效阻断下游通路,抑制肿瘤细胞增殖。另外由于爱必妥是IgG1的单克隆抗体,其具有Fc段功能,因此能结合免疫效应细胞活性的FcγRIII,通过免疫效应细胞杀伤靶细胞,即通过ADCC效应发挥抗肿瘤作用[9]。ADCC效应中爱必妥作为连接效应NK细胞与肿瘤细胞的桥梁发挥作用。
尽管爱必妥与化疗结合可将mCRC中位生存期延长至约24个月,但是单药有效率不高于10%,如何进一步提高疗效,克服原发和继发耐药成为临床研究的重要课题。研究表明,联合使用不同靶点的靶向治疗药物可能是逆转爱必妥耐药的有效策略之一。
BRAF是EGFR/RAS下游通路上重要基因,是一种重要的致癌基因。BRAF突变在CRC中的发生率约为8%~16%[11-12]。目前通过基因检测已明确BRAF突变的结直肠癌患者较野生型患者更容易发生复发转移且无进展生存期(PFS)和总生存期(OS)缩短。越来越多的证据支持,BRAF突变的晚期结直肠癌患者比野生型患者对爱必妥治疗的有效率低或根本无反应。然而BRAF突变是否可以作为EGFR单抗的疗效预测因子,目前国际癌症组织尚无统一定论[13-14]。本研究将浓度为0.1~100.0 μg/mL的爱必妥分别作用于BRAF突变型和野生型结直肠癌细胞株中48和72 h,结果显示爱必妥对BRAF突变细胞株的增殖抑制作用并不显著,因此判断BRAF突变结直肠癌细胞株对抗EGFR治疗耐药。而BRAF野生型结直肠癌细胞株对EGFR单抗治疗有反应,判定为相对敏感。
维罗非尼是一种新型口服有效TKI类 BRAF 激酶抑制剂,其作用机制为竞争性及可逆性地与 BRAFV600E激酶结合,抑制激酶活化,阻断MAPK通路,从而抑制肿瘤细胞的增殖[15]。大量的临床试验已经证实,维罗非尼对不可切除和转移性黑色素瘤治疗有效[16]。然而,在转移性BRAFV600E突变的结直肠癌中此类TKI药物的有效率还不足10%。研究证实,EGFR 和MAPK通路的反馈激活是BRAFV600E突变的结直肠癌患者对维罗非尼耐药的主要原因,在 BRAFV600E突变的结直肠癌患者中,联合使用EGFR 抑制剂与维罗非尼可提高抗肿瘤的疗效[17]。并且Martinelli等[18]发现多靶点的小分子TKI类靶向药物索拉非尼(多吉美)无论在BRAF突变或野生型细胞株中都能有效提高爱必妥疗效,这为本研究提供了理论依据。本研究将0.1~50.0 μg/mL 的维罗非尼分别作用于BRAF突变型和野生型结直肠癌细胞株48和72 h,结果显示维罗非尼对两种突变状态不同的结直肠癌细胞株均有增殖抑制作用,IC50均不高于7 μmol/mL,其在BRAF突变细胞株中更低。进一步选用单药维罗非尼(2 μmol/mL)、爱必妥(10 μg/mL)及两药联合作用72 h 后,与爱必妥单药作用组相比,细胞增殖抑制效果均得到加强,且差异有统计学意义,P<0.05。在长时间点集落形成实验中,两药联合对不同突变状态的结直肠癌细胞集落的形成也有明显的抑制作用。结果表明,无论在BRAF突变还是野生型细胞中,维罗非尼均能有效提高爱必妥作用的敏感性。
MAPKs 通路由一组级联激活的激酶组成,其激活是肿瘤发生、发展过程中的关键环节。ERK1/2 是 MAPK 通路重要的家族蛋白的成员之一,活化的ERK与转录因子相结合,在调节细胞周期、增殖、分化、细胞死亡、迁移等行为中发挥重要作用[19]。现已明确,细胞外信号通过激活EGFR并级联激活下游通路中的关键激酶来促进结直肠癌细胞的增殖、分化,参与肿瘤的发生、发展。作为这条通路中的关键因子,BRAF基因的激活突变使下游通路持续活化而导致肿瘤细胞不断分裂、增殖,最终发生转移。目前,以该通路中各个关键因子为靶点的抗肿瘤药物正在临床前及临床实验中广泛研究[20]。本研究结果显示,维罗非尼联合爱必妥能够加强其抑制结直肠癌细胞株增殖的效果,并且抑制EGFR、ERK的磷酸化,因此两药联合通过抑制 RAS/RAF/MEK/ERK 信号转导通路的各个环节,下调 p-ERK1/2 的表达,起到联合抗肿瘤作用。综上所述,本研究证实了无论在BRAFV600E突变还是BRAF野生型结直肠癌细胞中,维罗非尼均可增强爱必妥抗肿瘤活性,增效作用是通过联合作用,加强对EGFR及其下游信号转导通路的抑制而实现的。后续研究将在动物体内进行实验,验证细胞实验得到的结论。本研究结果为结直肠癌的联合靶向治疗提供新的方法和思路。
[1] Ciardiello F, Tortora G. EGFR antagonists in cancer treatment[J]. N Engl J Med,2008,358(11):1160-1174.
[2] Capdevila J,Elez E,Macarulla T,et al.Anti-epidermal growthfactor receptor monoclonal antibodies in cancer treatment[J].Cancer Treat Rev,2009,35(4): 354-363.
[3] Tang Y, Lou J, Alpaugh RK, et al. Regulation of antibody-dependent cellular cytotoxicity by IgG intrinsic and apparent affinity for target antigen[J]. Immunol, 2007, 179(5): 2815-2823.
[4] Jonker DJ,O’Callaghan CJ,Karapetis CS,et al.Cetuximab for the treatment of colorectal cancer[J]. N Engl J Med,2007,357(20): 2040-2048.
[5] You B,Chen EX.Anti-EGFR Monoclonal antibodies for treatment of colorectal cancers: development of cetuximab and panitumumab[J].J ClinPharmacol,2011,52(2): 128-155.
[6] Tiangui Huang, Michael Karsy, Jian Zhuge,et al. B-Raf and the inhibitors: from bench to bedside[J].Journal of Hematology & Oncology, 2013, 6:30.
[7] De Roock W,Claes B,Bernasconi D,et al.Effects of KRAS,BRAF,NRAS,and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastaticcolorectal cancer: a retrospective consortium analysis[J].LancetOncol,2010,11(8): 753-762.
[8] Prahallad A,Sun C,Huang S,et al.Unresponsiveness of colon cancer to BRAF(V600E)inhibition through feedback activationof EGFR[J]. Nature,2012,483(7387): 100-103.
[9] Kopetz S, Desai J, Chan E, et al. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors[J]. Clin. Oncol, 2010, 28(Suppl): a3534.
[10]Affolter K,Samowitz W,Tripp S,et al.BRAF V600E mutation detection by immunohistochemistry in colorectal carcinoma[J].Genes Chromosomes Cancer,2013,52(8): 748-752.
[11]Kimura H, Sakai K, Arao T, et al. Antibody-dependent cellular cytotoxicity of cetuximab against tumor cells with wild-type or mutant epidermal growth factor receptor[J]. Cancer Sci., 2007, 98: 1275-1280.
[12]Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer[J]. Nature, 2002, 417:949-954.
[13]Xu M, Xie Y, Ni S, et al. The latest therapeutic strategies after resistance tofirst generation epidermal growth factor receptor tyrosine kinase inhibitors(EGFR TKIs)in patients with non-small cell lung cancer (NSCLC)[J]. Ann Transl Med, 2015, 3(7): 96.
[14]Ohashi K, Sequist LV, Arcila ME, et al. Lung cancers with acquire dresistance to EGFR inhibitors occasionally harbor BRAF gene mutations butlack mutations in KRAS, NRAS, or MEK1[J]. Proc Natl Acad Sci USA, 2012,109(31): E2127-E2133.
[15]Garbe C, Abusaif S, Eigentler TK. Vemurafenib[J]. Recent Results Cancer Res, 2014, 201: 215-225.
[16]Choueiri TK, Cheville J, Palescandolo E, et al. BRAF mutations inmetanephric adenoma of the kidney[J]. EurUrol, 2012, 62(5): 917-922.
[17]Kurppa KJ, Caton J, Morgan PR, et al. High frequency of BRAF V600E mutations in ameloblastoma[J]. J Pathol, 2014, 232(5): 492-498.
[18]Martinelli E, Troiani T, Morgillo F, et al. Synergistic antitumor activity of sorafenib in combination with epidermal growth factor receptor inhibitors in colorectal and lung cancer cells[J]. Clin Cancer Res., 2010, 16(20):4990-5001.
[19]Wortzel I,Seger R.The ERK cascade:Distinct functions within various subcellular organelles[J].Genes Cancer,2011, 2(3):195-209.
[20]Wong KK. Recent Developments in Anti-Cancer Agents Targeting the Ras/Raf/MEK/ERK Pathway[J].Recent Patents on Anti-Cancer Drug Discovery, 2009, 4:28-35.
Effects of Cetuximab Combined with Vemurafenib on Colorectal Cancer Cell Lines
MA Rui, XU Ling, QU Xiu-juan, CHE Xiao-fang, GUO Tian-shu, LIU Yun-peng
(1stAffil.Hosp.,ChinaMed.Uni.,Shenyang110001)
The effects of cetuximabin combined with vemurafenib on wild type and mutant BRAF colorectal cancer cell lines were investigated. The cell proliferation was measured using MTT assay. The inhibitory effects of single and combination on cell proliferation were detected. Colony formation experiment: The inhibitory effect of different treatment factors on cell proliferation was confirmed by long-term culture of cells. The expression of EGFR and phosphor-EGFR and downstream pathway protein were determined by Western blotting. The results showed that for single drug therapy, BRAF wild group is relatively sensitive to cetuximab treatment. The inhibition of 100 μg/mL is 26%-40% in 72 h, whereas the BRAF mutation group of two cell lines was resistant to cetuximab. Both BRAF wild or mutant group cell lines were sensitive to vemurafenib, IC50 was less than 7 μmol/mL. The cell proliferative inhibition test showed the effect of two drug combinations were significantly lower than single drug cetuximab (P<0.05). Long time colony forming experiments confirmed that the combination in the area and the number of colony formation was significantly reduced. Western Blot experiment indicated that compared with single drug, two drug combination was significantly reduced p-EGFR, BRAF, p-ERK expression. The results showed that BRAF inhibitor vemurafenib can increase the sensitivity of cetuximab in either BRAF wild-type or mutant colorectal cancer cell lines.
cetuximab; vemurafenib; EGFR; BRAF; colorectal cancer
国家自然科学基金项目(81673025);辽宁省自然科学基金项目(2014021069)
马瑞 女,助教,硕士。研究方向为肿瘤学。Tel:024-83282312,E-mail:mary-414@163.com
* 通讯作者。男,教授,博士生导师。研究方向为肿瘤学。Tel: 024-83282312,E-mail:cmuliuyunpeng@hotmail.com
2017-01-29;
2017-04-05
Q939.9;R73
A
1005-7021(2017)02-0083-06
10.3969/j.issn.1005-7021.2017.02.014
