普通小麦穗部性状QTL分析
2017-05-15孙中沛刘天相左希亚赵璟琛王中华李春莲
孙中沛,刘天相,左希亚,赵璟琛,王中华,李春莲
(1.西北农林科技大学农学院/旱区作物逆境生物学国家重点实验室,陕西杨凌 712100;2.西北农林科技大学附属中学,陕西杨凌 712100)
普通小麦穗部性状QTL分析
孙中沛1,刘天相1,左希亚1,赵璟琛2,王中华1,李春莲1
(1.西北农林科技大学农学院/旱区作物逆境生物学国家重点实验室,陕西杨凌 712100;2.西北农林科技大学附属中学,陕西杨凌 712100)
为给小麦穗部性状标记辅助选择提供可供选择的分子标记,并进一步对小麦穗部相关性状QTL进行精细定位及相关基因克隆,利用普通小麦Heyne×Lakin杂交F2代单粒传获得的145个F6代重组自交系(recombinant inbred line,RIL)群体,构建了含有2 210个标记(2 068个SNP标记和142个SSR标记)的总长度为2 139.35 cM的遗传连锁图谱,并利用该图谱对小麦穗部性状(穗长、小穗数、穗密度)进行了QTL分析。结果表明,共检测出16个加性QTL,其中,与穗长相关的QTL有6个,分布在2A、2D、3B、4D、5A和7D染色体上,可解释表型变异7.58%~15.94%;与小穗数相关的QTL有4个,分布在1A、4A和7D染色体上,可解释表型变异7.28%~14.78%;与穗密度相关的QTL有6个,位于4D、5A和6B染色体上,可解释表型变异5.60%~20.06%。
小麦;RIL群体;遗传图谱;穗部性状;QTL
小麦是世界主要的粮食作物之一,但由于近几年种植业结构的调整,小麦种植面积大大减少,加之,小麦增产幅度的下降,使得小麦总产量降低[1],这引起了全世界小麦领域专家的高度关注。小麦产量与小麦穗部性状高度相关[2],因此,研究小麦穗部性状将对于提高小麦产量具有重要意义。
迄今,已有许多国内外学者在不同的遗传背景及环境下,对产量以及穗部相关状进行了QTL定位研究。如,Schlegel等[3]将与小穗数相关的QTL定位在1BS染色体上;Börner等[4]利用普通小麦Opata85/W7984构建的重组自交系(recombinant inbred line, RIL)群体,检测到64个籽粒产量相关性状的QTL。Li等[5]利用RIL群体,将产量相关性状(千粒重、穗粒数、单株穗数、小穗数)的QTL定位在了小麦12条染色体上,并检测到了4个相关QTL 富集区,分别位于1D、2A、6B和7D染色体上;Huang等[6]利用BC2F1群体,将2个与单株穗数相关的QTL定位在了1B和7A染色体上;Kumar等[7]利用RIL群体对小麦的穗长、穗粒数、粒重、生物产量、收获指数等穗部性状进行了QTL定位,结果在2B和2D染色体上定位到2个控制穗长的QTL,在2B、4A和6A染色体上定位到3个控制每穗小穗数的QTL;Marza等[8]利用Ning 7840×Clark构建的包含132个家系的F12代RIL群体,在2BL、2BS、3BL、4B、5B、7A和7BS检测到与穗长相关的QTL。但是,由于穗部相关性状具有复杂的遗传背景,受环境因素的影响较大,导致不同研究者得到的结果差异较大,限制了其在育种中的应用。本研究利用源自普通小麦Heyne×Lakin的RIL群体构建了基于SNP和SSR标记的高密度遗传图谱,并结合在两年两点的环境中获得的表型数据进行小麦穗部相关性状的QTL定位,以期为小麦穗部性状标记辅助选择提供可供选择的分子标记,并为小麦穗部相关性状QTL的精细定位和基因克隆奠定基础。
1 材料与方法
1.1 供试材料
供试材料为普通冬小麦品种Heyne和Lakin及其二者杂交F2代通过单粒传获得的145个F6代RIL群体。Heyne对小麦锈病及赤霉病具有抗性,但产量相对较低;Lakin为感病品种,大粒,产量相对较高。
1.2 田间种植和穗部性状调查
所有供试材料于2015年和2016年在陕西省杨凌和三原两个地点进行种植。采用完全随机区组设计,2次重复,2行区,行长2 m,行距20 cm,株距5 cm,常规栽培管理。
穗部性状包括穗长、小穗数和穗密度。在小麦拔节期之后,观察其抽穗时期及开花时期,以亲本或每个株系抽穗(或开花)的植株数达到一半的时间为抽穗期(或开花期)。开花期过后,穗长已基本定型,取5个小麦主茎进行穗长的测定,然后取平均值。小麦成熟后,每个亲本或株系随机取5穗,考察小穗数,然后取平均值。穗密度=小穗数/穗长。
1.3 标记分析与高密度遗传连锁图谱的构建
采用CTAB法提取亲本及RIL群体的DNA。利用9K Illumina iSelect SNP 基因芯片技术及小麦全基因组SSR标记技术对由Heyne与Lakin构建的包含145个家系的F6代RIL群体进行分析,与母本Heyne相同的带型标记为2,与父本Lakin带型相同的标记为0,缺失带型标记为-1,最终共检测到2 068个SNP多态性标记和172个SSR多态性标记。
利用QTL IciMapping 4.0作图软件中的MAP模块对2 240个标记基因型进行连锁分析,使用Kosambi作图函数将重组率转换为遗传距离(cM)。
1.4 数据分析
结合RIL群体各株系植株的穗部性状表型数据和高密度遗传连锁图谱,利用QTL IciMapping 4.0软件以完备区间作图法(ICIM)[9]检测了控制小麦各穗部性状的QTL位点、分析了其染色体位置及加性遗传效应及贡献率。QTL检测中LOD阈值为2.5,逐步回归概率为P<0.001,步进区间为1.0 cM。
2 结果与分析
2.1 亲本及其RIL群体穗部相关性状的变异
根据两年两点的田间穗部性状调查结果(表1)可知,两亲本穗部相关性状表现出较大差异,亲本Heyne的小穗数变化范围为15.0~18.6个,穗长变化范围为9.10~10.62 cm,穗密度变化范围为1.57~1.84 个·cm-1;亲本Lakin的小穗数变化范围为15.2~20.4个,穗长为8.90~11.58 cm,穗密度为1.57~1.91 个·cm-1。
同样,RIL群体内穗部相关性状也表现出较大差异,差异达极显著水平(P<0.001),且表现为连续变异,小穗数变化范围为11.7~22.9个,穗长变化范围为6.85~17.20 cm,穗密度变化范围为1.28~2.30 个·cm-1。W检验结果均在0.9以上,表明RIL群体在各环境中穗部相关性状呈近似正态分布。
2.2 遗传连锁图谱的构建及分析
连锁遗传分析结果表明,共有2 210个标记(2 068个SNP标记和142个SSR标记)覆盖了小麦全部21条染色体。连锁遗传图谱的总长度为2 139.35 cM,两个标记间的平均距离为0.96 cM。标记数、遗传长度及标记密度在小麦三个染色体组间存在着明显差异,但在同源群间的差异相对较小,具体结果见表2。
A、B、D分别表示A、B、D染色体组;1~7分别表示1~7同源染色体群。
A,B and D indicated gomome A,gonome B and gonome C,respectively;1-7 indicated homologous chromosome groups 1-7,respectively.
2.3 穗部性状的QTL定位及遗传分析
对RIL群体小麦穗部性状进行了QTL分析,共检测到16个加性QTL,分别位于小麦的1A、2A、2D、3B、4A、4D、5A、6B和7D染色体上(表3、图1)。其中,在2A、2D、3B、4D、5A、7D染色体上定位到6个穗长QTL,解释的表型变异率为7.58%~15.94%, Qsl-3B的贡献率最高(15.94%),标记区间为Xwmc78-IWA6464。 Qsl-2A、 Qsl-2D、 Qsl-3B、 Qsl-7D增加穗长的等位基因来源于亲本Heyne, Qsl-4D、 Qsl-5A增加穗长的等位基因来源于亲本Lakin。4个小穗数QTL被定位于1A、4A、7D染色体上,其中 Qsn-1A.1、 Qsn-1A.2、 Qsn-7D增加小穗数的等位基因来源于亲本Heyne, Qsn-4A增加小穗数的等位基因来源于亲本Lakin。这些QTL解释的表型变异率为7.28%~14.78%,其中 Qsn-4A贡献率最高(14.78%),标记区间为Xbarc343-IWA6690。 Qsn-1A.2在三个环境中均检测到,为稳定的QTL,标记区间为IWA3374-IWA1724,标记间距离为0.73 cM。6个穗密度QTL被定位于4D、5A和6B染色体上,其中,5A染色体上共检测到4个加性效应QTL( Qsc-5A.1、 Qsc-5A.2、 Qsc-5A.3和 Qsc-5A.4)。各QTL解释的表型变异率为5.60%~20.06%, Qsc-5A.2的贡献率最高,标记区间为IWA825-IWA3530。 Qsc-4D、 Qsc-5A.1、 Qsc-5A.2和 Qsc-5A.3的加性效应来源于Heyne, Qsc-5A.4和 Qsc-6B的加性效应来源于Lakin。
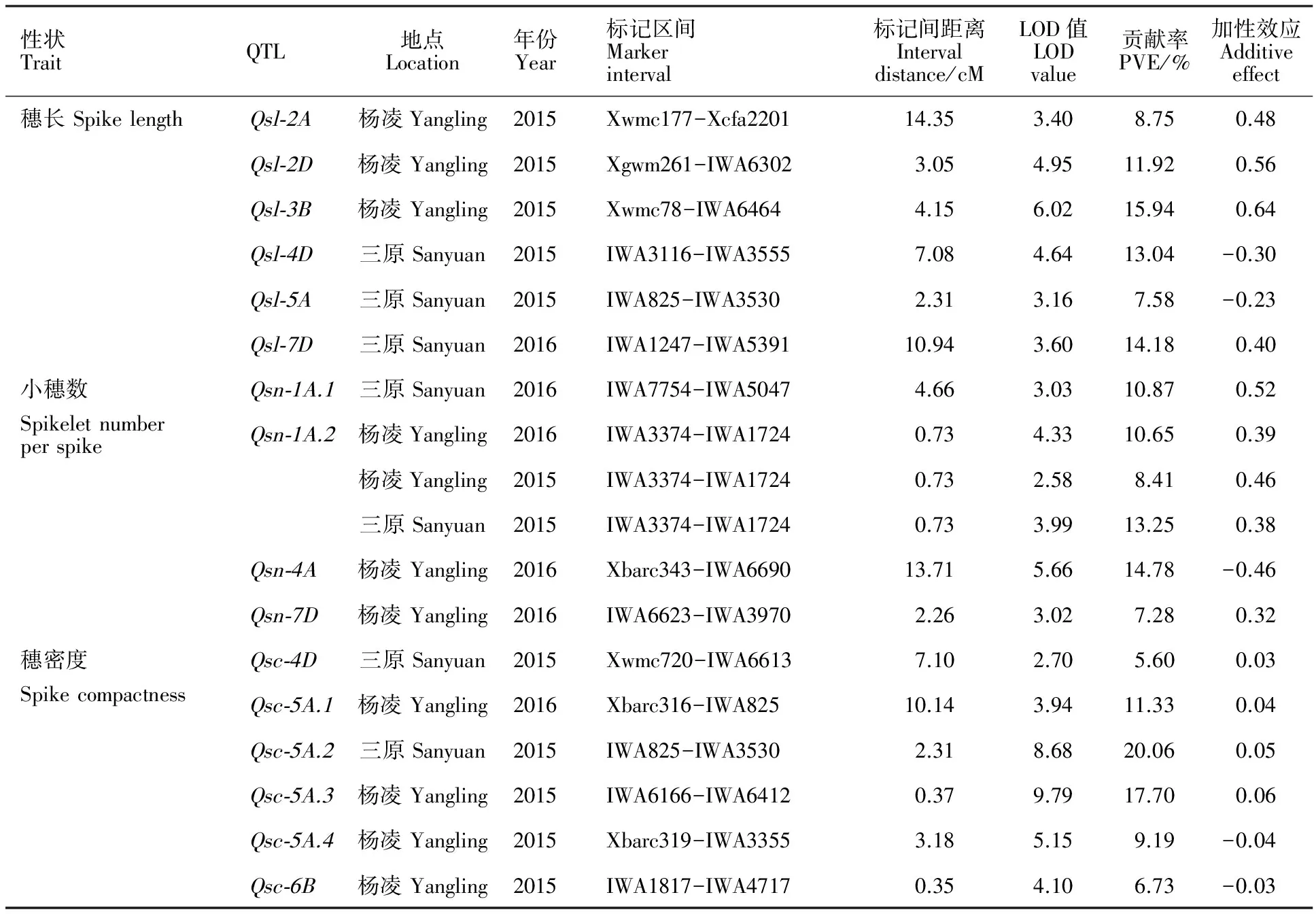
表3 穗部相关性状QTL定位结果
3 讨 论
Ma等[10]利用RIL群体对穗部相关性状进行了QTL定位,在1A、2D、4A、5A、5B和7D染色体上检测到控制穗长的QTL,在1B、2D、3B、5A、5B和7D染色体上检测到控制小穗数的QTL。其中,控制穗长的位点 QSpl.nau-2D,标记区间为Xgwm26-XRPP5,与本研究所检测到的控制穗长的位点 Qsl-2D位置相同,说明这两个控制穗长的QTL位点是同一位点。此外,Ma等[10]检测到的另一个控制穗长的位点 QSpl.nau-5A,标记区间为Xwmc96-Xgwm304,与本研究所检测到的控制穗长的位点 Qsl-5A遗传距离仅为6.33 cM,说明本研究在5A染色体上发现的控制穗长的位点 Qsl-5A与Ma等[10]检测到的位点 QSpl.nau-5A紧密连锁。因此,这两个控制穗长的QTL位点很可能是同一位点。Sourdi-lle等[11]利用DH群体,在不同年份不同地点种植的环境条件下,使用单因素方差分析法对小麦的农艺性状进行单标记QTL检测,在2D染色体上检测到一个与小麦穗长具有高度相关性的分子标记Xgwm261,这与本研究定位到一个控制穗长的位点 Qsl-2D,标记区间Xgwm261-IWA6302,所得结果一致。通过与前人研究的比较可知本研究定位到的位点 Qsl-2D和 Qsl-5A是能够在不同作图群体以及不同环境下都能检测到的稳定的QTL位点。
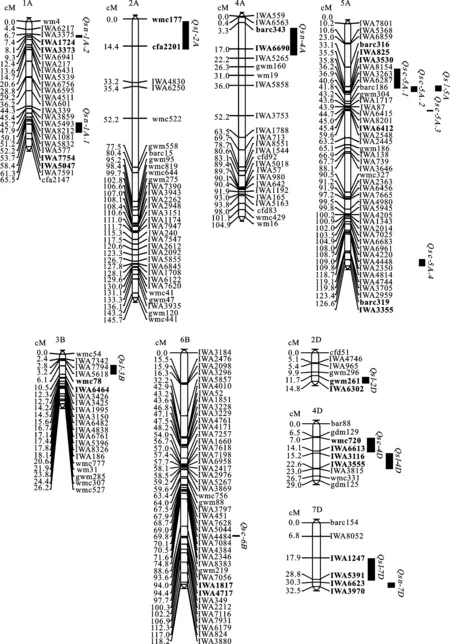
图1 小麦穗部相关性状的加性QTL在染色体上的分布
张坤普等[12]利用小麦品种花培3号和豫麦57杂交获得的DH群体也对穗部性状进行了QTL研究,在2B、2D、4D、5D、6B染色体上检测到控制穗长的QTL;在1B、4A、5D和7A染色体上检测到控制小穗数的QTL;在2D、4D和5D染色体上检测到控制穗密度的QTL。其中,一个控制穗密度的位点 qSc-2D,标记区间为Xgwm261-Xgwm296,与本研究检测到控制穗长的位点 Qsl-2D位置相同。说明标记Xgwm261附近区段可能对于控制穗长和穗密度两个性状均发挥作用,由于穗密度=小穗数/穗长,因此某遗传区段影响穗长的同时也很有可能影响穗密度。值得注意的是,在本研究中根据穗长和穗密度检测到的QTL位点 Qsl-5A(IWA825-IWA3530)和 Qsc-5A.2(IWA825-IWA3530)均被定为于5A染色体上,且位置相同,这说明控制穗长与穗密度性状的基因有可能是同一位点。
众多的小麦穗部性状QTL研究中,利用不同的作图群体检测出同一个性状QTL的位置并不是完全相同的,这些差异表明,小麦穗部性状大都属于数量性状遗传,遗传基础较为复杂,与遗传环境条件和背景等密切相关[13]。所以不同研究中QTL定位结果的差异是客观存在的。通过获得更多更准确的表型数据,以及进行分子标记加密,可使定位结果更为可靠和准确。本研究所得的结果为分子标记辅助选择和基因聚合育种提供了可供选择的分子标记,并为小麦穗部相关性状的精细定位和基因克隆等进一步深入研究奠定了基础。
[1] 于振文,田奇卓,潘庆明,等.黄淮麦区冬小麦超高产栽培的理论与实践[J].作物学报,2002,28(5):577.
YU Z W,TIAN Q Z,PAN Q M,etal.Theory and practice on cultivation of super high yield of winter wheat in the wheat fields of Yellow River and Huaihe River districts [J].ActaAgronomicaSinica,2002,28(5):577.
[2] 魏艳丽,王彬龙,李瑞国,等.大穗小麦穗部性状的遗传分析[J].麦类作物学报,2015,35(10):1366.
WEI Y L,WANG B L,LI R G,etal.Genttic analysis on spike characteristics of wheat variety with large spike [J].JournalofTriticeaeCrops,2015,35(10):1366.
[3]SCHLEGEL R,MEINEL A.A quantitative trait locus(QTL) on chromosome arm 1RS of rye and its effect on yield performance of hexaploid wheat [J].CerealResearchCommunications,1994,22:7.
[4]BÖRNER A,SCHUMANN E,FURSTE A.Mapping of quantitative trait loci determining agronomic important characters in hexaploid wheat(TriticunaestivumL.) [J].TheoreticalandAppliedGenetics,2002,105:921.
[5]LI S S,JIA J Z,WEI X Y,etal.A intervarietal genetic map and QTL analysis for yield traits in wheat [J].MolecularGeneticsandGenomics,2007,20:167.
[6]HUANG X Q,COSTER H,GANAL M W,etal.Advanced backcross QTL analysis for identification of quantitative trait loci alleles from wild relatives of wheat(TriticumaestivumL.) [J].TheoreticalandAppliedGenetics,2003,106:1379.
[7]KUMAR N,KULWAL P L,BALYAN H S.QTL mapping for yield and yield contributing traits in two mapping populations of bread wheat [J].MolecularBreeding,2007,19(2):163.
[9]MARZA F,BAI G H,CARVER B F,etal.Quantitative trait locus for yield and related traits in the wheat population Ning 7840×Clark [J].TheoreticalandAppliedGenetics,2006,112:688.
[9]LI H H,YE G Y,WANG J K,etal.A modified algorithm for the improvement of composite interval mapping [J].GeneticsSocietyofAmerica,2007,175:361.
[10]MA Z Q,ZHAO D M,ZHANG C Q,etal.Molecular genetic analysis of five spike-related traits in wheat using RIL and immortalized F2population [J].MolecularGeneticsandGenomics,2007,277:31.
[11]SOURDILLE P,CADALEN T,GUYOMARC'H H.An update of the Courtot×Chinese Spring intervarietal molecular marker linkage map for the QTL detection of agronomic traits in wheat [J].TheoreticalandAppliedGenetics,2003,106:530.
[12] 张坤普,徐宪斌,田纪春.小麦籽粒产量及穗部相关性状的QTL定位[J].作物学报,2009,35(2):270.
ZHANG K P,XU X B,TIAN J C.QTL mapping for grain yield and spike related traits in common wheat [J].ActaAgronomicaSinica,2009,35(2):270.
[13] 常 鑫,李法计,张兆萍,等.小麦旗叶长、宽及面积的QTL分析[J].西北植物学报,2014,34(5):896.
CHANG X,LI F J,ZHANG Z P,etal.Mapping QTLs for flag leaf length,width and area in wheat [J].ActaBotanicaBoreali-OccidentaliaSinica,2014,34(5):896.
QTL Mapping of Spike-Related Traits in Common Wheat
SUN Zhongpei1,LIU Tianxiang1,ZUO Xiya1,ZHAO Jingchen2,WANG Zhonghua1,LI Chunlian1
(1.College of Agronomy,Northwest A&F University/State Key Laboratory of Crop Stress Biology for Arid Areas,Yangling, Shaanxi 712100,China;2.High School Attached to Northwest A&F University,Yangling,Shaanxi 712100,China)
Wheat is one of the most important crops in the world, and the spike-related traits of wheat are closely related to the yield of wheat. Thus, it is of great significance to study the spike related traits of wheat. This study characterized quantitative trait loci (QTL) underlying wheat spike-related traits using a high-density genetic linkage map developed from a recombinant inbred line (RIL) population derived from a cross between 'Heyne' and 'Lakin'. The map consists of 2 068 single nucleotide polymorphism (SNP) and 142 simple sequence repeat (SSR) markers covering a genetic distance of 2 139.35 cM. The RIL population was evaluated for spike length (SL), spikelet number per spike (SPN), and spike compactness (SC) at Yangling and Sanyuan. Using QTL mapping, sixteen additive QTLs for spike-related traits were detected, including six of them for SL were mapped on chromosomes 2A, 2D, 3B, 4D, 5A and 7D, explaining 7.58% to 15.94% of the phenotypic variances; four QTLs for SPN were located on chromosomes 1A, 4A and 7D, accounting for 7.28% to 14.78% of the phenotypic variances;six QTLs for spike compactness (SC) were mapped on chromosomes 4D, 5A and 6B, explaining the phenotypic variation of 5.6% to 20.06%. The findings shed light on the inheritance of spike-related traits and provide DNA markers for marker-assisted selection in wheat breeding.
Wheat; RIL population; Genetic map;Spike-related traits; QTL
时间:2017-04-07
2017-01-27
2017-03-11 基金项目:2017西北农林科技大学唐仲英作物育种基金项目;陕西省重点科技创新团队项目(2014KCT-25) 第一作者E-mail:sunzhongpei@126.com 通讯作者:李春莲(E-mail:lclian@163.com)
S512.1;S336
A
1009-1041(2017)04-0452-07
网络出版地址:http://kns.cnki.net/kcms/detail/61.1359.S.20170407.1020.008.html
