Recent development and gene therapy for glycogen storage disease type Ia☆
2017-05-07JaniceChouGooYoungKimJunHoCho
Janice Y.Chou,Goo-Young Kim,Jun-Ho Cho
Section on Cellular Differentiation,Eunice Kennedy Shriver National Institute of Child Health and Human Development,National Institutes of Health,Bethesda,MD,USA
1.Introduction
Glycogen storage disease type I(GSD-I),also known as von Gierke disease,consists oftwo major subtypes,GSD-Ia(MIM232200),caused by a de ficiency in glucose-6-phosphatase-α(G6Pase-αor G6PC)and GSD-Ib(MIM232220),caused by a de ficiency in the glucose-6-phosphate(G6P)transporter(G6PT).1-3The incidence of GSD-I is approximately 1 in 100,000,with GSD-Ia being the most prevalent form,representing approximately 80%of cases.G6Pase-αcatalyzes the hydrolysis of G6P to glucose and phosphate in the terminal steps of gluconeogenesis and glycogenolysis,and is primarily expressed in the liver,kidney,and intestine.1-3The active site of G6Pase-αlies on the luminal side of the endoplasmic reticulum(ER),inaccessible to the cytoplasm.4Therefore,for G6P hydrolysis to occurin vivo,the G6P substrate must be translocated from the cytoplasm into the ER lumen by the G6PT.1-3Accordingly,G6P transport and hydrolysis are tightly coupled events and the primary function of the G6Pase-α/G6PT complex is to maintain interprandial blood glucose homeostasis.1-3Patients affected by GSD-Ia are unable to maintain glucose homeostasis and present with fasting hypoglycemia,hepatomegaly,nephromegaly,hyperlipidemia,hyperuricemia,lactic acidemia,and growth retardation.1-3GSD-Ia is lethal in childhood,if untreated.Dietary therapies have enabled GSD-Ia patients to attain near normal growth and pubertal development.5,6However,no current therapy is able to address long-term complications of hepatocellular adenoma(HCA)that develop in 75%of GSD-I patients over 25 years-old.1-3,7,8In 10%of cases,HCA undergoes malignant transformation to hepatocellular carcinoma(HCC).1-3,8,9This review focuses on recent developments in GSD-Ia,including gene therapy for the treatment of this disorder.
2.Molecular genetics of GSD-Ia
2.1.The G6PC gene and encoded G6Pase-αenzyme
HumanG6PCis a single copy gene composed of five exons(Fig.1A)located on chromosome 17q21.10The encoded G6Pase-α enzyme is a 357 amino-acid glycoprotein anchored to the ER membrane by nine transmembrane helices(Fig.1B).10-12Based on mutational and active site labeling studies,the current paradigm for the G6Pase-αreaction mechanism is that His-176 initiates a nucleophilic attack on the phosphate of G6P to form a phosphohistidine-G6Pase-αintermediate.4This transition state is stabilized by Arg-83 hydrogen bonding to phosphate,and is resolved by His-119 providing a proton that liberates the glucose moiety.The active site residues of G6Pase-α,Arg-83,His-119,and His-176 are all situated inside the ER lumen(Fig.1B),inaccessible to G6P in the cytoplasm.
To date,95 separateG6PCmutations(Fig.2),including 63 missense,10 nonsense,17 insertion/deletion,4 splicing,and 1 nostop mutation(c.1074A>C/p.358Yext43)have been identi fied[http://www.hgmd.cf.ac.uk/ac/gene.php?gene=G6PC]. Of the identi fiedG6PCmutations,51 missense(Fig.3),2 nonsense(p.R170X and p.Q347X), and 2 codon deletion(c.734_735insG743delC andp.F327del)mutationshavebeen con firmed as pathogenic based on site-directed mutagenesis and transient expression assays.13-15Thirty-three of theG6PCmissense mutations completely abolish G6Pase-αactivity,and the other 18 retain varying degrees of residual enzymatic activity.13-15Mutations in two of the proposed G6Pase-αactive site residues,namely p.R83C,p.R83H,and p.H119L(Fig.3),have been identi fied in GSD-Ia patients and shown to completely abolish G6Pase-αenzymatic activity,consistent with their proposed role.10,14No mutation has been found in the phosphate acceptor p.H176 in G6Pase-α.However,site-directed mutagenesis has shown that the p.H176A mutation completely abolished G6Pase-αenzymatic activity.16Four natural occurringG6PCsplicing mutations,c.230+4A>G,c.231-1G>A,c.563-3C>G,and c.648G>T have been identi fied(Fig.2).The c.231-1G>A mutation causes exon 2 skipping and the c.648G>T mutation results in a 91-nt deletion in exon 5 encoding a severely truncated polypeptide of 201 amino acids.17-19Both mutations are predicted to inactivate G6Pase-αactivity.While GSD-Ia is not predominantly restricted to any one racial or ethnic group,mutations in theG6PCgene unique to Caucasian,Hispanic,Chinese/Japanese/Korean,and Jewish GSD-Ia patients have been described,suggestingseparateethnicfoundereffectsforsomemutations.1,2,20,21GSD-Ia is more prevalent in the Ashkenazi Jewish population,where the carrier frequency for the p.R83C mutation is 1.4%.20To date,no clear genotype-phenotype correlations have been demonstrated for GSD-Ia.2,21
2.2.The metabolic phenotype of GSD-Ia
The hallmark of GSD-Ia is hypoglycemia following a short fast of a few hours.The liver,and to a lesser extent,the kidney and intestine,are the primary gluconeogenic organs involved in the regulation of blood glucose homeostasis between meals.As blood glucose levels fall between meals,G6P produced in the terminal step of gluconeogenesis and glycogenolysis in the gluconeogenic organs is transported by G6PT into the ER,where G6P is hydrolyzed by G6Pase-αto glucose for release back into the blood(Fig.4).The defective G6Pase-αenzyme in the liver,kidney,and intestine of GSD-Ia patients results in impaired blood glucose homeostasis(Fig.5).1-3Associated with this is an elevation of G6P in the cytoplasm of the cells,leading to an excessive accumulation of glycogen,which promotes progressive hepatomegaly and nephromegaly seen in GSD-Ia patients.Hepatomegaly is further exacerbated byan accumulation of neutral lipids in the liver.Other major metabolic consequences of elevated liver and kidney cytoplasmic G6P are hypercholesterolemia,hypertriglyceridemia,hyperuricemia,and lactic acidemia that characterize the clinical pathophysiology of GSD-Ia(Fig.5).Longer-term presentations of GSD-Ia include growth retardation,osteoporosis,gout,pulmonary hypertension,HCA with risk of transformation to HCC,and renal disease.1-3
3.GSD-Ia animal models
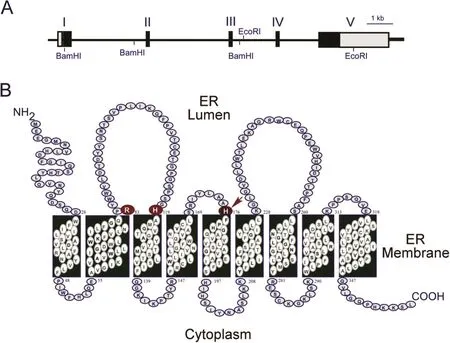
Fig.1.The G6PC gene and the encoded G6Pase-αenzyme.(A)G6PC,located on human chromosome 17q21,is shown as a line diagram with exons marked as boxes I to V.Black boxes represent coding regions,white boxes the 5′and 3′untranslated regions of the G6PC transcript.(B)The encoded G6Pase-α enzyme is anchored in the membrane of the ER by nine transmembrane helices.The amino-terminus is located in the ER lumen and the carboxyl-terminus in the cellular cytoplasm.Arg-83,His-119,and His-176,which contribute to the active center,are denoted by large circles.The phosphate acceptor,His-176,is denoted by an arrow.
Several GSD-Ia animal models exist,including a globalG6pcknock-out mouse model(G6pc-/-),22a naturally occurring dog model,23and two conditionalG6pc-null mouse models.24,25TheG6pc-/-mouse model manifests all known symptoms of human GSD-Ia,making it an excellent model of the human disease.22However,G6pc-/-mice exhibit relatively mild lactic acidemia compared with human GSD-Ia patients.The GSD-Ia dog model is based on a naturally occurring p.M121IG6PCmutation identi fied in the Maltese breed that was cross-bred with beagles to overcome size,neonatal survival,and small litter size limitations of the carrier Maltese background.23This Maltese-Beagle hybrid GSD-Ia dog manifests all of the typical symptoms of the human disorder,including lactic acidosis typical of human GSD-Ia patients.These animal models have been widely used to understand the biology,pathophysiology,and long-term complications of GSD-Ia,and have been utilized to develop gene and cell therapies for GSD-Ia.26,27TheG6pc-/-mice andG6pc-/-dogs rarely survive longer than three weeks,even under intensive dietary therapy regimes.22,23Consequently,HCA development has not been reported in the untreated globalG6pcknockout animal models.However,liver-specificG6pcnull(L-G6pc-/-)mice survive to adulthood and develop multiple HCA,25,28which can be used to study the etiology of HCA/HCC associated with GSD-Ia.In addition,kidney-specificG6pcknockout and intestine-specific knockout mice are available to delineate molecular mechanisms underlying other long-term complications of GSD-Ia.29,30

Fig.2.Mutations identi fied in the G6PC gene of GSD-Ia patients.G6PC is shown as a line diagram with exons marked as boxes I to V.Black boxes represent coding regions,white boxes the 5′and 3′untranslated regions of the G6PC transcript.The positions of all known mutations are listed from left to right as insertion/deletion(black),nonsense(red),splicing(green),and missense(blue)mutations.
4.GSD-I-associated HCA
4.1.Hepatic tumors identi fi ed in human GSD-I
One severe long-term complication of GSD-I is HCA,a benign liver tumor that develops in 75%of GSD-I patients over 25 yearsold.1-3HCAsin GSD-Ipatientsare small,multiple,nonencapsulated,and produce excess hepcidin that contributes to anemia.31Additional complications from GSD-I-associated HCA include intratumoral hemorrhage and,in 10%of cases,malignant transformation to HCC can occur.Previously,molecular analysis classi fied HCA into four major subgroups:hepatocyte nuclear factor 1A mutated HCA(HHCA),in flammatory HCA(IHCA),β-catenin(CTNNB1exon 3)mutated HCA(bex3HCA),and unclassi fied HCA(UHCA).32More recently,a new HCA subgroup,shHCA,caused by activation of sonic hedgehog signaling,was identi fied.33shHCA represents 4%of all HCAs,and is associated with obesity and bleeding.HCAs are now classi fied into eight subgroups:HHCA,IHCA,bex3HCA with an increased risk of malignant transformation to HCC,bex7,8HCA without an increased risk of malignant transformation,bex3IHCA,bex7,8IHCA,shHCA,and UHCA.33The bex3HCA and bex7,8HCA areβ-catenin-mutated HCAs inCTNNB1exon 3 and exon 7 or 8,respectively.The bex3IHCA and bex7,8IHCA exhibited both in flammatory phenotype and β-catenin activating mutations.33
Calderaroet al.34characterized 25 HCAs that developed in 14 GSD-Ia and 1 GSD-Ib patients using gene expression and DNA sequence of mutated genes in sporadic HCAs.They classi fied GSD HCAs as IHCA(52%),bHCA(28%),or UHCA(20%).The bHCA seen in GSD-Ia patients were characterized by high expression of twoβcatenin target genes,glutamate-ammonia ligase(GLUL),and leucine-rich repeat containing G protein-coupled receptor 5(LGR5).34The IHCA were characterized by a constitutive activation of signal transducer and activatorof transcription(STAT3),a cancerpromoting transcription factor.34,35Interestingly,while HHCA was the major sporadic HCA identi fied,no HHCA was observed in GSD-I patients.34Calderaroet al.34showed that GSD-I livers without tumors and the HHCA exhibited similar metabolic defects characterized by gluconeogenesis repression,glycolysis activation,and fatty acid synthesis activation,providing one clue why HHCA had not been identi fied in GSD-I patients.Based on the new HCA classi fication,33the seven bHCA cases identi fied in human GSD-I patients are:bex3HCA(n=3),bex3IHCA(n=1),bex7,8HCA(n=2),and bex7,8IHCA(n=1).Presently,6 HCA subgroups(IHCA,bex3HCA,bex3IHCA,bex7,8HCA,bex7,8IHCA,and UHCA)have been identi fied in GSD-I patients.The underlying etiology of HCA in GSD-Ia is unknown.
4.2.Hepatic tumors identi fi ed in murine GSD-Ia
Using magnetic resonance imaging,Mutelet al.25showed that LG6pc-/-mice developed hepatic nodules nine months after gene deletion and all mice developed multiple HCAs 18 months after gene deletion,but they did not characterize the tumor subtypes.Of the three rAAV-treatedG6pc-/-mice that developed hepatic tumors at age 71-72 weeks,one harbored a HCA nodule and the other two each harbored a single HCC lesion.36The non-tumor liver regions of the two HCC-bearing rAAV-treatedG6pc-/-mice had 1.5-2.2 units of G6Pase-αactivity.However,the HCC lesions of the rAAV-treatedG6pc-/-mice had non-detectable G6Pase-αactivity.36HCC-1 expressed increased levels of mRNA forGlulandLgr5,suggesting that HCC-1 was derived from a bHCA.HCC-2 displayed increased levels of the active p-STAT3-Y705 protein,suggesting that HCC-2 was derived from an IHCA.36
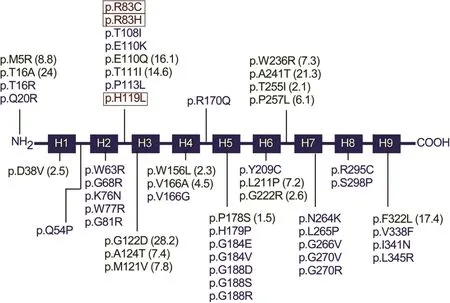
Fig.3.A summary of missense protein mutations identi fied in human GSD-Ia patients that affect phosphohydrolase activity.The G6Pase-αprotein is represented by a line diagram,with the nine helical transmembrane domains marked as boxes H1 to H9.Protein mutations that destroy G6Pase-αactivity are listed in blue.Mutants retaining some residual activity are listed in green with the percent of wild-type enzymatic activity retained in parentheses.Mutations identi fied in the active site residues that destroy enzymatic activity are listed in red and bracketed.
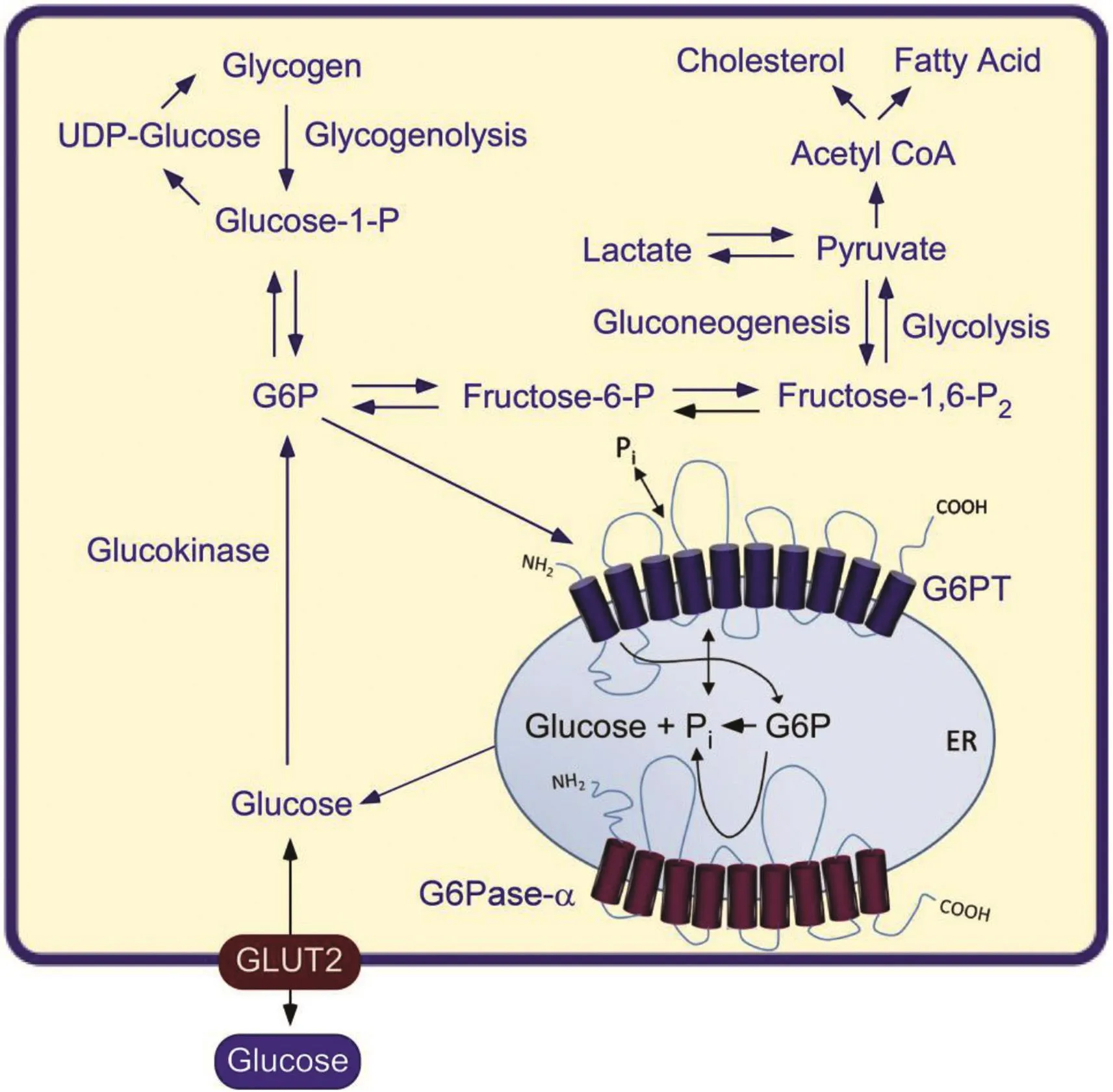
Fig.4.The primary anabolic and catabolic pathways of G6P in the liver,kidney,and intestine.Intracellular glucose is produced via hydrolysis of G6P by G6Pase-αin the terminal rate-limiting step of gluconeogenesis and glycogenolysis.The G6Pase-α and G6PTcomponents of the G6Pase-α/G6PTcomplex are shown embedded within the membrane of the ER.The GLUT2 transporter,responsible for the transport of glucose in and out of the cell,is shown embedded in the plasma membrane.Abbreviations:G6P,glucose-6-phosphate;UDP-Glucose,uridine 5′-diphosphate glucose.
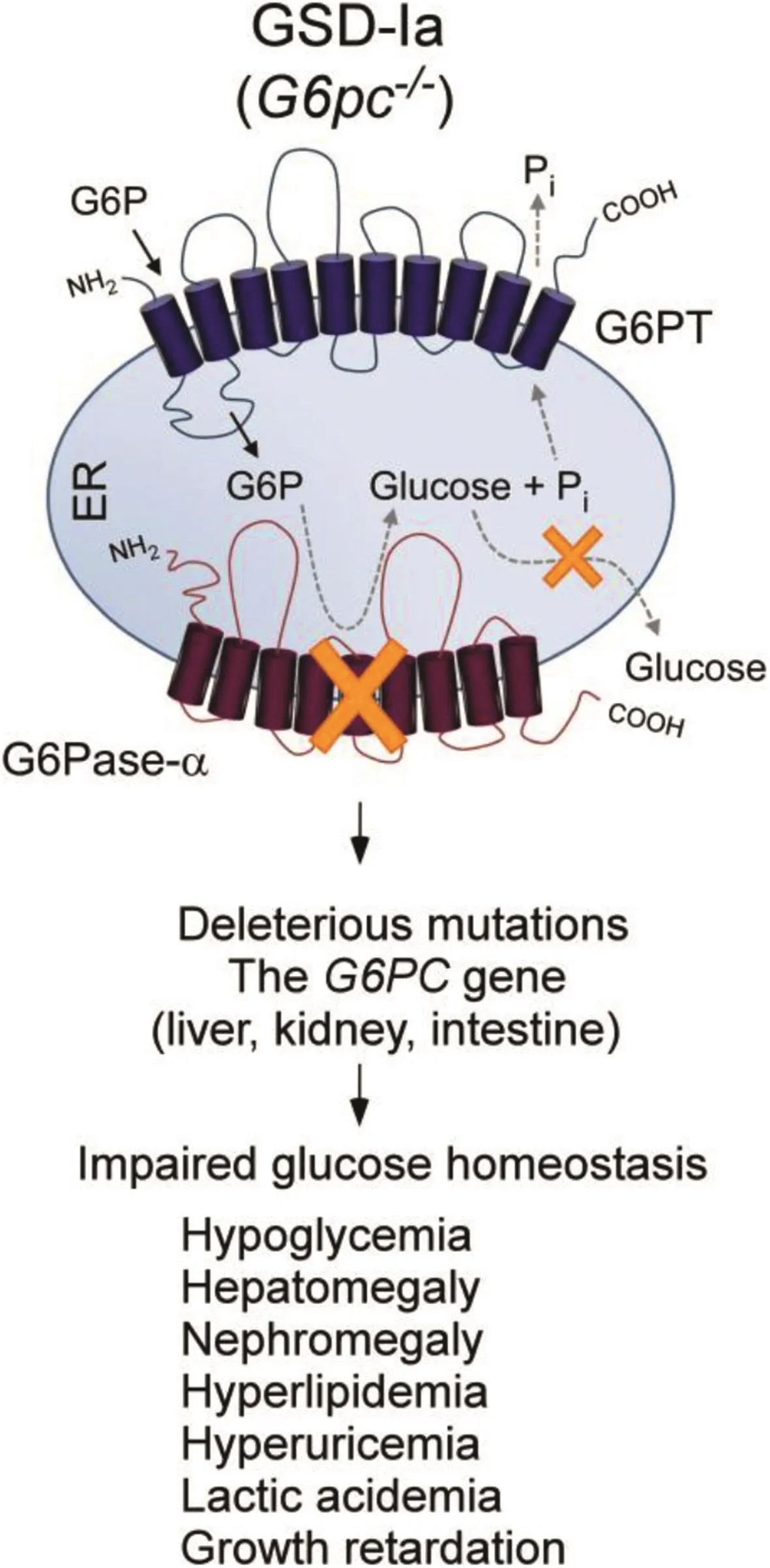
Fig.5.GSD-Ia is caused by deleterious mutations in the G6PC gene and manifests a phenotype of impaired glucose homeostasis.The G6Pase-αand G6PT components of the G6Pase-α/G6PT complex are shown embedded within the membrane of the ER.Abbreviation:G6PC,Glucose-6-phosphatase-α.
5.Treatment
5.1.Conventional treatment
Metabolicdisruption presentin GSD-Iapatientscan be adequately managed with strong adherence to dietary therapies.1-3GSD-Ia infants typically receive nocturnal nasogastric infusion of glucose to avoid hypoglycemia.5GSD-Ia patients 3 years or older are prescribed uncooked cornstarch,a slow release carbohydrate,to prolong the length of euglycemia between meals.6These dietary therapies enable GSD-Ia patients to maintain normoglycemia,but the underlying pathological processes remain uncorrected.Consequently,HCA/HCC and renal disease,two major causes of morbidity and mortality in patients with GSD-Ia,are common.1-3
5.2.Gene therapy
G6Pase-αis an extremely hydrophobic transmembrane protein that is dif ficult to purify and protein replacement therapy is not an option,11,12but somatic gene therapy is a promising approach.A variety of gene transfer vectors,including adenovirus vectors,37helper-dependent adenovirus vectors,38lentivirus vectors,39-41and recombinant adeno-associated virus (rAAV)vectors,26,27,36,42-47have been developed using animal models of GSD-Ia.The rAAV-mediated genetherapy in both mouse and canine models of GSD-Ia has led to long-term correction of metabolic abnormalities with no detectable toxicity.36,42-47The most promising results come from GSD-Ia studies using rAAV vectors directed by the humanG6PCpromoter/enhancer(GPE).43,44rAAV8-miGPE and rAAV8-GPE are human G6Pase-α-expressing rAAV2/8 vectors driven by 382-bp of the minimal(mi)GPE(rAAV8-miGPE)and approximately 3 kb GPE(rAAV8-GPE),43,44respectively.Both vectors demonstrated ef ficacy in treatingG6pc-/-mice and the rAAV8-miGPE vector also showed ef ficacy in GSD-Ia dogs.43-45While a cell-mediated immune response of hepatic CD8+lymphocyte in filtration was observed inG6pc-/-mice infused with a G6Pase-αexpressing rAAV8 vector driven by the CBA/CMA promoter/enhancer,no such immune response was observed with rAAV8-GPE under the same conditions.44
To select the best vector for clinical translation,a direct comparison of rAAV8-GPE and rAAV8-miGPE vectors was conducted usingG6pc-/-mice.46The results show that the rAAV8-GPE vector directs significantly higher levels of hepatic G6Pase-αexpression,achieves greater reduction in hepatic glycogen accumulation,and leads to a better tolerance of fasting,than the rAAV8-miGPE vector.46This suggests that the rAAV8-GPE vector is the best vector for clinical translation in human GSD-Ia.In a long-term dose-ranging study,Leeet al.45further showed that rAAV8-GPE-mediated gene transfer showed that restoring≥3%(5 units)of normal hepatic G6Pase-αactivity inG6pc-/-mice was suf ficient to maintain glucose homeostasis.The treatedG6pc-/-mice displayed normal hepatic fat storage,normal blood metabolite and glucose tolerance pro files,reduced fasting blood insulin levels,and had no evidence of hepatic abnormalities or HCA.Fasting hypoglycemia is the hallmark of GSD-Ia.Promisingly,the rAAV8-GPE-treatedG6pc-/-mice were able to sustain a 24-h fast,which is a stress test of the liver's ability to maintain blood normoglycemia through glycogenolysis and gluconeogenesis catalyzed by G6Pase-αin the absence of dietary glucose(Fig.6).Leeet al.45further showed that the ability of rAAV-GPE-treatedG6pc-/-mice expressing low levels of hepatic G6Pase-αto produce suf ficient glucose to maintain interprandial glucose homeostasis correlated with an increase in hepatic G6PT mRNA expression and a corresponding increase in microsomal G6P uptake activity(Fig.6).
More recently,Kimet al.36conducted a second long-range study to examine the minimum dosage of rAAV-GPE vector required to prevent HCA formation inG6pc-/-mice.The authors characterized 11 rAAV-GPE-treatedG6pc-/-mice harboring 0.9%-2.4%(1.5-4.1 units)of normal hepatic G6Pase-αactivity,and showed that three mice expressing 0.9%-1.3%of normal hepatic G6Pase-αactivity developed HCA/HCC and eight did not.The results established a threshold of hepatic G6Pase-αactivity required to prevent HCA/HCC,and showed that GSD-Ia mice harboring less than 2%of normalhepatic G6Pase-α activity are at risk of tumor development.36
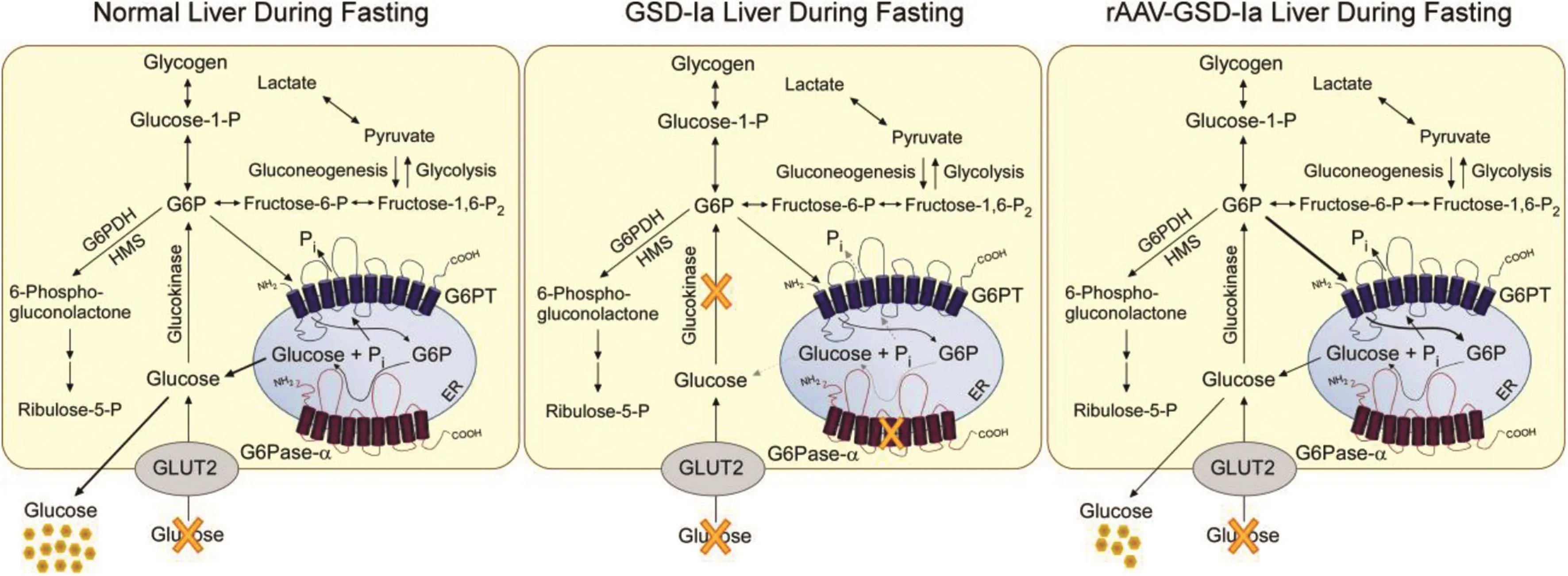
Fig.6.Pathways for G6P metabolism in the livers of normal,GSD-Ia,and rAAV8-GPE-treated GSD-Ia mice during fasting.During fasting,G6P,the end product of gluconeogenesis and glycogenolysis,is transported from the cytoplasm into the lumen of the ER by G6PT.Inside the ER,G6P is hydrolyzed by G6Pase-αand the resulting glucose is transported back into the cytoplasm then released into circulation.In the GSD-Ia liver,which lacks a functional G6Pase-α,the ER-localized G6P cannot be converted to glucose,leading to hypoglycemia following a short fast.The rAAV8-GPE-treated GSD-Ia(rAAV-GSD-Ia)liver,which expresses reduced levels of G6Pase-α,but increased levels of G6PT compared with a normal liver,generates reduced levels of endogenous glucose and maintains interprandial glucose homeostasis.The GLUT2 transporter,responsible for the transport of glucose in and out of the cell,is shown embedded in the plasma membrane.G6PT and G6Pase-αare shown embedded in the ER membrane.
Correction of renal disease in GSD-Ia has been less extensively studied.rAAV8-mediated gene transfer results in little or no renal G6Pase-αexpression,and the abnormal renal pathology persists.This has been attributed to poor kidney transduction mediated by the AAV2/8 serotype.43,46,48Different AAV serotypes have different tissue transduction ef ficiencies and more recent data suggest that rAAV2/9 may be the preferred choice for future renal gene delivery.49-52However,rAAV2/9-mediated transgene expression in the kidney is still significantly lower than that in the liver.Using a retrograde renal vein injection method,Roccaet al.52showed that rAAV2/9-mediated kidney transduction could be markedly improved.Identi fication of viral serotypes that effectively transduce all affected tissue types remains to be further explored.Notably,serotypes can have very different targeting ef ficiencies in different species.Only a small number of serotypes have been used in clinical trials to date.Therefore,there is a need to understand more about the primate specificity of the many serotypes that appear promising in rodents.
6.Conclusions
Metabolic abnormalities in GSD-Ia are currently being treated by dietary therapies that have enabled patients to maintain euglycemia and remove early symptomatic signs of the disease.However,dietary therapies leave the patient vulnerable to severe long-term complications of renal disease and HCA/HCC.The effective use of gene therapies to correct the disease in GSD-Ia animal models is very promising,with efforts to initiate clinical trials on the horizon.While early interventions of gene therapy prevent HCA development,it is unclear whether gene therapy can abrogate preexisting hepatic tumors.Delineating the molecular mechanisms underlying HCA/HCC in GSD-Ia remains to be explored.The LG6pc-/-mice that survive to adulthood and develop HCA/HCC offer a suitable model to study the etiology of hepatic tumors and their treatment of GSD-Ia.It is important to identify potential targets to guide the development of therapies,not only to correct metabolic abnormalities,but to eradicate the long-term complications of this disorder.
Con flict of interest
The authors declare that they have no con flict of interest.
Acknowledgements
This research was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development,National Institutes of Health(HD000912-38).
1.Chou JY,Matern D,Mans field BC,Chen YT.Type I glycogen storage diseases:disorders of the glucose-6-phosphatase complex.Curr Mol Med.2002;2:121-143.
2.Chou JY,Jun HS,Mans field BC.Glycogen storage disease type I and G6Pase-β de ficiency:etiology and therapy.Nat Rev Endocrinol.2010;6:676-688.
3.Chou JY,Jun HS,Mans field BC.Type I glycogen storage diseases:disorders of the glucose-6-phosphatase/glucose-6-phosphate transporter complexes.J Inherit Metab Dis.2015;38:511-519.
4.Ghosh A,Shieh JJ,Pan CJ,Sun MS,Chou JY.The catalytic center of glucose-6-phosphatase:His176is the nucleophile forming the phosphohistidine-enzyme intermediate during catalysis.J Biol Chem.2002;277:32837-32842.
5.Greene HL,Slonim AE,O'Neill Jr JA,Burr IM.Continuous nocturnal intragastric feeding for management of type 1 glycogen-storage disease.N Engl J Med.1976;294:423-425.
6.Chen YT,Cornblath M,Sidbury JB.Cornstarch therapy in type I glycogenstorage disease.N Engl J Med.1984;310:171-175.
7.Labrune P,Trioche P,Duvaltier I,Chevalier P,Odiˋevre M.Hepatocellular adenomas in glycogen storage disease type I and III:a series of 43 patients and review of the literature.J Pediatr Gastroenterol Nutr.1997;24:276-279.
8.Rake JP,Visser G,Labrune P,Leonard JV,Ullrich K,Smit GP.Glycogen storage disease type I:diagnosis,management,clinical course and outcome.Results of the European Study on Glycogen Storage Disease Type I(ESGSD I).Eur J Pediatr.2002;161:S20-S34.
9.Franco LM,Krishnamurthy V,Bali D,et al.Hepatocellular carcinoma in glycogen storage disease type Ia:a case series.J Inherit Metab Dis.2005;28:153-162.
10.Lei KJ,Shelly LL,Pan CJ,Sidbury JB,Chou JY.Mutations in the glucose-6-phosphatase gene that cause glycogen storage disease type 1a.Science.1993;262:580-583.
11.Pan CJ,Lei KJ,Annabi B,Hemrika W,Chou JY.Transmembrane topology of glucose-6-phosphatase.J Biol Chem.1998;273:6144-6148.
12.Pan CJ,Lei KJ,Chou JY.Asparagine-linked oligosaccharides are localized to a luminal hydrophilic loop in human glucose-6-phosphatase.J Biol Chem.1998;273:21658-21662.
13.Chou JY,Mans field BC.Mutations in the glucose-6-phosphatase-alpha(G6PC)gene that cause type Ia glycogen storage disease.Hum Mutat.2008;29:921-930.
14.Shieh JJ,Terizioglu M,Hiraiwa H,et al.The molecular basis of glycogen storage disease type 1a:structure and function analysis of mutations in glucose-6-phosphatase.J Biol Chem.2002;277:5047-5053.
15.Shieh JJ,Lu YH,Huang SW,et al.Misdiagnosis as steatohepatitis in a family with mild glycogen storage disease type 1a.Gene.2012;509:154-157.
16.Lei KJ1,Pan CJ,Liu JL,Shelly LL,Chou JY.Structure-function analysis of human glucose-6-phosphatase,the enzyme de ficient in glycogen storage disease type 1a.J Biol Chem.1995;270:11882-11886.
17.Akanuma J,Nishigaki T,Fujii K,et al.Glycogen storage disease type Ia:molecular diagnosis of 51 Japanese patients and characterization of slicing mutations by analysis of ectopically transcribed mRNA from lymphoblastoid cells.Am J Med Genet.2000;91:107-112.
18.Kajihara S,Matsuhashi S,Yamamoto K,et al.Exon rede finition by a point mutation within exon 5 of the glucose-6-phosphatase gene is the major cause of glycogen storage disease type 1a in Japan.Am J Hum Genet.1995;57:549-555.
19.Okubo M,Aoyama Y,Kishimoto M,Shishiba Y,Murase T.Identi fication of a point mutation(G727T)in the glucose-6-phosphatase gene in Japanese patients with glycogen storage disease type 1a,and carrier screening in healthy volunteers.Clin Genet.1997;51:179-183.
20.Ekstein J,Rubin BY,Anderson SL,et al.Mutation frequencies for glycogen storage disease Ia in the Ashkenazi Jewish population.Am J Med Genet A.2004;129A:162-164.
21.Matern D,Seydewitz HH,Bali D,Lang C,Chen YT.Glycogen storage disease type I:diagnosis and phenotype/genotype correlation.EurJPediat.2002;161(Suppl 1):S10-S19.
22.Lei KJ,Chen H,Pan CJ,et al.Glucose-6-phosphatase dependent substrate transport in the glycogen storage disease type-1a mouse.Nat Genet.1996;13:203-209.
23.Kishnani PS,Bao Y,Wu JY,Brix AE,Lin JL,Chen YT.Isolation and nucleotide sequence of canine glucose-6-phosphatase mRNA:identi fication of mutation in puppies with glycogen storage disease type 1a.Biochem Mol Med.1997;61:168-177.
24.Peng WT,Pan CJ,Lee EJ,Westphal H,Chou JY.Generation of mice with a conditional allele for G6pc.Genesis.2009;47:590-594.
25.Mutel E,Abdul-Wahed A,Ramamonjisoa N,et al.Targeted deletion of liver glucose-6 phosphatase mimics glycogen storage disease type 1a including development of multiple adenomas.J Hepatol.2011;54:529-537.
26.Chou JY,Mans field BC.Gene therapy for type I glycogen storage diseases.Curr Gene Ther.2007;7:79-88.
27.Chou JY,Mans field BC.Recombinant AAV-directed gene therapy for type I glycogen storage diseases.Expert Opin Biol Ther.2011;11:1011-1024.
28.Resaz R,Vanni C,Segalerba D,et al.Development of hepatocellular adenomas and carcinomas in mice with liver-specific G6Pase-αde ficiency.Dis Model Mech.2014;7:1083-1091.
29.Clar J,Gri B,Calderaro J,et al.Targeted deletion of kidney glucose-6 phosphatase leads to nephropathy.Kidney Int.2014;86:747-756.
30.Rajas F,Clar J,Gautier-Stein A,Mithieux G.Lessons from new mouse models of glycogen storage disease type 1a in relation to the time course and organ specificity of the disease.J Inherit Metab Dis.2015;38:521-527.
31.Weinstein DA,Roy CN,Fleming MD,Loda MF,Wolfsdorf JI,Andrews NC.Inappropriate expression of hepcidin is associated with iron refractory anemia:implications for the anemia of chronic disease.Blood.2002;100:3776-3781.
32.Bioulac-Sage P,Laumonier H,Couchy G,et al.Hepatocellular adenoma management and phenotypic classi fication:the Bordeaux experience.Hepatology.2009;50:481-489.
33.Nault JC,Couchy G,Balabaud C,et al.Molecular classi fication of hepatocellular adenoma associates with risk factors,bleeding,and malignant transformation.Gastroenterology.2017;152:880-894.
34.Calderaro J,Labrune P,Morcrette G,et al.Molecular characterization of hepatocellular adenomas developed in patients with glycogen storage disease type I.J Hepatol.2013;58:350-357.
35.Pilati C,Zucman-Rossi J.Mutations leading to constitutive active gp130/JAK1/STAT3 pathway.Cytokine Growth Factor Rev.2015;26:499-506.
36.Kim GY,Lee YM,Kwon JH,et al.Glycogen storage disease type Ia mice with less than 2%of normal hepatic glucose-6-phosphatase-αactivity restored are at risk of developing hepatic tumors.Mol Genet Metab.2017;120:229-234.
37.Zingone A,Hiraiwa H,Pan CJ,et al.Correction of glycogen storage disease type 1a in a mouse model by gene therapy.J Biol Chem.2000;275:828-832.
38.Crane B,Luo X,Demaster A,et al.Rescue administration of a helper-dependent adenovirus vector with long-term ef ficacy in dogs with glycogen storage disease type Ia.Gene Ther.2012;19:443-452.
39.Salani B,Damonte P,Zingone A,et al.Newborn liver gene transfer by an HIV-2-based lentiviral vector.Gene Ther.2005;12:803-814.
40.Grinshpun A,Condiotti R,Waddington SN,et al.Neonatal gene therapy of glycogen storage disease type Ia using a feline immunode ficiency virus based vector.Mol Ther.2010;18:1592-1598.
41.Clar J,Mutel E,Gri B,et al.Hepatic lentiviral gene transfer prevents the longterm onset of hepatic tumours of glycogen storage disease type 1a in mice.Hum Mol Genet.2015;24:2287-2296.
42.Ghosh A,Allamarvdasht M,Pan CJ,et al.Long-term correction of murine glycogen storage disease type Ia by recombinant adeno-associated virus-1-mediated gene transfer.Gene Ther.2006;13:321-329.
43.Koeberl DD,Pinto C,Sun B,et al.AAV vector-mediated reversal of hypoglycemia in canine and murine glycogen storage disease type Ia.Mol Ther.2008;16:665-672.
44.Yiu WH,Lee YM,Peng WT,et al.Complete normalization of hepatic G6PC de ficiency in murine glycogen storage disease type Ia using gene therapy.Mol Ther.2010;18:1076-1084.
45.Lee YM,Jun HS,Pan CJ,et al.Prevention of hepatocellular adenoma and correction of metabolic abnormalities in murine glycogen storage disease type Ia by gene therapy.Hepatology.2012;56:1719-1729.
46.Lee YM,Pan CJ,Koeberl DD,Mans field BC,Chou JY.The Upstream enhancer elements of the G6PC promoter are critical for optimal G6PC expression in murine glycogen storage disease type Ia.Mol Genet Metab.2013;110:275-280.
47.Lee YM,Kim GY,Pan CJ.Mans field BC,Chou JY.Minimal hepatic glucose-6-phosphatase-αactivity required to sustain survival and prevent hepatocellular adenoma formation in murine glycogen storage disease type Ia.Mol Genet Metab Rep.2015;3:28-32.
48.Akache B,Grimm D,Pandey K,et al.The 37/67-kilodalton laminin receptor is a receptor for adeno-associated virus serotypes 8,2,3,and 9.J Virol.2006;80:9831-9836.
49.Michelfelder S,Trepel M.Adeno-associated viral vectors and their redirection to cell-type specific receptors.Adv Genet.2009;67:29-60.
50.Zincarelli C,Soltys S,Rengo G,Rabinowitz JE.Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection.Mol Ther.2008;16:1073-1080.
51.Schievenbusch S,Strack I,Schef fler M,et al.Combined paracrine and endocrine AAV9 mediated expression of hepatocyte growth factor for the treatment of renal fibrosis.Mol Ther.2010;18:1302-1309.
52.Rocca CJ,Ur SN,Harrison F,Cherqui S.rAAV9 combined with renal vein injection is optimal for kidney-targeted gene delivery:conclusion of a comparative study.Gene Ther.2014;21:618-628.
杂志排行

Liver Research的其它文章
- Interleukin-22 in the pathogenesis and potential treatment of liver diseases☆
- FOXO transcription factors in non-alcoholic fatty liver disease☆
- Long non-coding RNA in liver metabolism and disease:Current status☆
- Interaction between stress responses and circadian metabolism in metabolic disease☆
- Decoding the role of extracellular vesicles in liver diseases☆
- Guide for Authors