ABT-263中间体羧酸片段合成研究
2017-04-27申焕玲周国春
申焕玲, 周国春
(南京工业大学药学院, 南京210009)
ABT-263中间体羧酸片段合成研究
申焕玲, 周国春
(南京工业大学药学院, 南京210009)
报道了Bcl-2家族蛋白抑制剂ABT-263(1)中间体羧酸片段即4-(4-((2-(4-氯苯基)-5,5-二甲基环己基-1-烯)甲基)哌嗪-1-基)苯甲酸(2)的合成方法。首先4,4-二甲基环己酮与三溴化磷和N,N-二甲基甲酰胺反应生成第一个中间体化合物2-溴-5,5-二甲基环己基-1-烯甲醛(3),经Suzuki偶联反应,还原、卤代、胺化,最后经水解、酸化得到目标产物(2)。经过6步反应,使用廉价易得的原料和试剂,反应条件温和,后处理方便,纯化方法简单,总产率达到48.0%,明显高于现有文献报道产率,适合大规模生产。各步反应产物结构经过1H NMR确认。
ABT-263;羧酸片段;合成
引言
细胞凋亡或程序性细胞死亡是通过消除损坏和老化的细胞而保持机体正常的发育和组织的完整性[1-3]。在这个调节过程中的任何改变都将引发包括癌症在内的一系列疾病。Bcl-2家族蛋白是细胞凋亡的关键调节因子,在调节细胞凋亡信号中发挥重要作用[4-6]。因此,将Bcl-2家族蛋白作为潜在的治疗药物靶点成为现今抗癌药物研究的热点,进而研发出一类前景较好的抗癌化合物[7-8](图1)。Wang[9-15]课题组报道了一系列化合物,如棉子酚[9]、TW-37[10]、TM-1206[12]等;René Grée[16-18]课题组也报道了一些化合物,如三唑衍生物[16]等;雅培制药(Abbott Laboratories)[19-21]报道了ABT-737[19]、ABT-263[20-21](1)等活性化合物。虽然这些化合物都存在口服生物利用度低,给药困难等问题,但研究表明ABT-263口服生物利用度高于同类型的其他活性化合物,因此受到了广泛的关注。
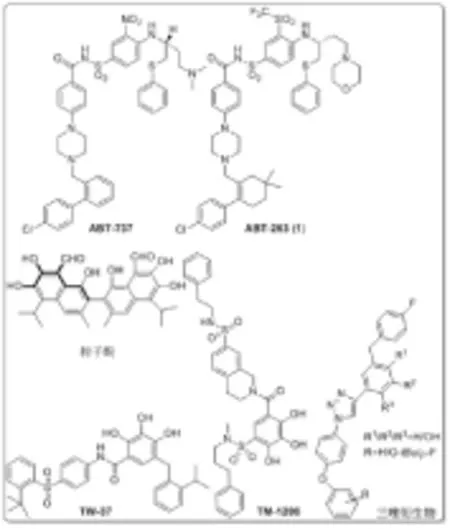
图1以Bcl-2家族蛋白为药物靶点的重要分子
ABT-263的逆合成分析[20](图2)显示化合物(1)由羧酸部分化合物(2)和苯磺酰胺部分两个中间体缩合而成,本文的研究目的是改进ABT-263羧酸部分化合物(2)的合成方法。自化合物(2)被报道以来,近几年合成方法主要有雅培和药明康德两条合成路线。在雅培制药公司合成化合物(2)的过程中[20],还原胺化产率只有38.0%,并且使用了毒性较大的氰基硼氢化钠,总收率只达到了14.0%,而且大多中间产物分离纯化困难。药明康德公司的合成路线[22],由于溶剂残留特别是水份对下一步卤代反应会有较明显的不良影响,而醛和醇的沸点都较低(实验研究发现在油泵真空干燥时醛和醇在5小时内分别减重85.4%和70.2%左右),因此在大规模生产过程中产品的干燥可能比较困难;另外,该工艺中胺烷基化和偶联反应的总收率只有62.0%,而且偶联反应需要三种混合溶剂,虽然使用贵金属催化剂的偶联反应放在后面合成阶段,成本上较为合理,但胺的偶联中用到了大量的较贵的碘化钠(3.04eq),这样也使成本上升。与雅培制药公司工艺相比虽然产率提高了,但是仍然有较多不足之处。
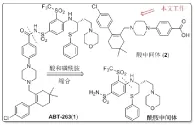
图2ABT-263逆合成分析
综合以上两条合成路线[20-22],本文提出以4,4-二甲基环己酮为原料,与三溴化磷和N,N-二甲基甲酰胺反应合成化合物(3),经过简单处理的化合物(3)(前一步的溶剂残留对下一步反应几乎没有影响)可以直接进行Suzuki偶联反应,然后进行醛还原、烯丙位伯醇氯代、胺烷基化,最后酯水解和酸化得到终产物(2)(图3)。
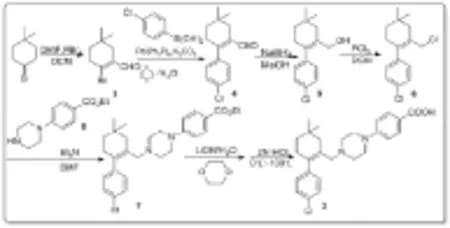
图3化合物(2)的合成路线
1实验部分
1.1试剂与仪器
DZF-6050真空干燥器(上海一恒科学仪器有限公司);旋转蒸发器RE-5299,DF-101S型集热式恒温加热磁力搅拌器(巩义市予华仪器有限责任公司);Bruker-400 MHz型核磁共振仪(瑞士Bruker公司);Agilent LS/MS型高分辨质谱议(HRMS)(美国Agilent公司)。
4-(哌嗪-1-基)苯甲酸乙酯(8)按照文献[23]方法合成;4,4-二甲基环己酮(试剂级,安微济达化工有限公司);四三苯基磷钯(试剂级,萨恩化学技术(上海)有限公司);三溴化磷、哌嗪、4-氟苯甲酸乙酯、4-氯苯硼酸(试剂级,基丽化学技术(上海)有限公司);N,N-二甲基甲酰胺、二氯甲烷、乙酸乙酯、甲醇、1,4-二氧六环、石油醚(分析纯,西陇化工股份有限公司);其他无机试剂均为分析纯,上海凌峰化学试剂有限公司。
1.2合成
1.2.14-(哌嗪-1-基)苯甲酸乙酯(8)的合成
称取哌嗪12.29 g(142.7 mmol)和K2CO313.15 g(95.14 mmol)置于250 mL的三口烧瓶中,N2保护,加入150 mL二甲基亚砜。预冷,缓慢加入4-氟苯甲酸乙酯8.0 g(47.57 mmol)。滴加完毕后,升温至120 ℃。搅拌6 h,TLC监测反应无原料,结束反应。冷却至室温,将体系缓慢加入到冷水中,乙酸乙酯萃取多次,依次用蒸馏水、饱和NaHCO3水溶液、饱和NaCl水溶液洗涤两次,有机相用无水Na2SO4干燥,过滤,滤液浓缩后得化合物(8)[23]10.48 g,产率为94.1%。1H NMR(400 MHz,(CD3)2SO):δ7.77(d,J=9.1 Hz,2H,4,6-H),6.95(d,J=9.1 Hz,2H,1,3-H),4.23(q,J=7.1 Hz,2H,17-H),3.19~3.22(m,4H,8,12-H),2.79~2.81(m,4H,9,11-H),1.28(t,J=7.1 Hz,3H,16-H)。无需纯化,直接投下一步。
1.2.22-溴-5,5-二甲基环己基-1-烯甲醛(3)的合成
N2保护,0 ℃下,将20.20 mL三溴化磷(216.34 mmol)缓慢滴加到150 mL无水二氯甲烷和18.80 mL无水N,N-二甲基甲酰胺(246.18 mmol)的混合溶液中,滴加完毕后,体系慢慢恢复至室温,并在此温度下搅拌0.5 h。再次冷却至0 ℃,将4,4-二甲基环己酮9.40 g(74.60 mmol)的80 mL无水无氧二氯甲烷溶液缓慢滴入到上述体系中,滴加完毕后,慢慢恢复至室温,此时加上尾气吸收处理装置,并搅拌过夜。TLC监测反应无原料,结束反应。将反应液缓慢倒入到冰冷的饱和NaHCO3水溶液中,石油醚萃取,后处理方式与1.2.1相同。浓缩后得黄色油状液体化合物(3)[20-22]10.47 g,产率65.0%。1H NMR(400 MHz,CDCl3):δ10.01(s,1H,10-H),2.73~2.76(m,2H,2-H),2.07(s,2H,5-H),1.51(t,J=6.5 Hz,2H,1-H),0.93(s,6H,7,8-H)。无需纯化,直接投下一步。
1.2.32-对氯苯基-5,5-二甲基环己基-1-烯甲醛(4)的合成
N2保护,0 ℃下, 将干燥后的3 10.47 g(48.47 mmol)的145 mL 1,4-二氧六环溶液缓慢加入到4-氯苯硼酸7.58 g(48.47 mmol)与四三苯基磷钯151 mg (0.13 mmol)的混合物中,滴加完毕,升温至100 ℃,小心分批加入145 mL K2CO313.38 g(96.94 mmol)水溶液,继续搅拌2 h,TLC监测反应无原料,结束反应。体系冷却至室温,乙酸乙酯萃取,后处理方式与1.2.1相同。过滤,滤液浓缩后用硅胶柱层析纯化(石油醚/乙酸乙酯=15/1)得黄色油状液体化合物(4)[24]10.34 g,产率86.0%。1H NMR (400 MHz,CDCl3):δ9.49(s,1H,16-H),7.36(d,J=8.5 Hz,2H,12,14-H),7.16(d,J=8.5 Hz,2H,11,15-H),2.52~2.56(m,2H,2-H),2.14(s,2H,5-H),1.53(t,J=6.4 Hz,2H,1-H),0.99(s,6H,7,8-H)。
1.2.4(2-对氯苯基-5,5-二甲基环己基-1-烯)甲醇(5)的合成
0 ℃下,将化合物(4)10.34 g(41.69 mmol)溶于125 mL甲醇中,加入硼氢化钠1.90 g(50.03 mmol)。保持0 ℃,搅拌2 h。TLC监测反应无原料,结束反应,将体系缓慢倒入冰水中,搅拌0.5 h,乙酸乙酯萃取,后处理方式与1.2.1相同。浓缩后得淡黄色油状液体化合物(5)[22,25-26]10.42 g,产率100%。1H NMR(400 MHz,(CD3)2SO):δ7.36(d,J=8.4 Hz,2H,12,14-H),7.21(d,J=8.4 Hz,2H,11,15-H),4.52(t,J=5.3 Hz,1H,17-H),3.68(d,J=5.2 Hz,2H,10-H),2.23(t,J=5.9 Hz,2H,2-H),1.98(s,2H,5-H),1.41(t,J=6.4 Hz,2H,1-H),0.95(s,6H,7,8-H)。无需纯化,直接投下一步。
1.2.5(2-对氯苯基-5,5-二甲基环己基-1-烯)氯甲烷(6)的合成
0 ℃下,将化合物(5) 10.42 g(41.68 mmol)溶于125 mL无水二氯甲烷,然后快速称量并加入五氯化磷4.33 g(20.84 mmol)。搅拌4 h,TLC监测反应无原料,结束反应。将体系缓慢倒入冰水中,搅拌至澄清透明,乙酸乙酯萃取,后处理方式与1.2.1相同。浓缩后得无色液体化合物(6)[22,26]10.52 g,产率为94.2%。1H NMR (400 MHz,CDCl3):δ 7.32 (d,J=8.39 Hz,2H,12,14-H),7.16(d,J=8.39 Hz,2H,11,15-H),3.88(s,2H,10-H),2.30(t,J=6.38 Hz,2H,2-H),2.05(s,2H,5-H),1.48(t,J=6.46 Hz,2H,1-H),1.00(s,6H,7,8-H)。无需纯化,直接投下一步。
1.2.64-(4-((2-(4-氯苯基)-5,5-二甲基环己基-1-烯)甲基)哌嗪-1-基)苯甲酸乙酯(7)的合成
常温下,将化合物(6)10.52 g(39.25 mmol)、化合物(8)9.185 g(39.25 mmol)溶于118 mLN,N-二甲基甲酰胺,缓慢加入三乙胺13.68 mL(98.12 mmol),升温至80 ℃。搅拌3 h,TLC监测反应无原料,结束反应。将体系缓慢倒入冰水中,搅拌至澄清透明,乙酸乙酯萃取,后处理方式与1.2.1相同。浓缩后得白色固体化合物(7)[22,26]17.56 g,产率96.0%。1H NMR(400 MHz,CDCl3):δ7.89(d,J=9.0 Hz,2H,25,27-H),7.28(d,J=1.7 Hz,2H,12,14-H),7.00(d,J=8.4 Hz,2H,11,15-H),6.81(d,J=9.0 Hz,2H,24,28-H),4.32(q,J=7.1 Hz,2H,33-H),3.25~3.27(m,4H, 19,21-H),2.80(s,2H,10-H),2.34~2.37(m,4H,18,22-H),2.26(t,J=5.3 Hz,2H,2-H),2.02(s,2H,5-H),1.47(t,J=6.5 Hz,2H,1-H),1.36(t,J=7.1 Hz,3H,32-H),0.99(s,6H,7,8-H)。无需纯化,直接投下一步。
1.2.74-(4-((2-(4-氯苯基)-5,5-二甲基环己基-1-烯)甲基)哌嗪-1-基)苯甲酸(2)的合成
将化合物(7)17.56 g(37.68 mmol)溶于435 mL 1,4-二氧六环。预冷,缓慢将87 mL 1N氢氧化锂水溶液滴加至上述体系,升温至100 ℃。搅拌24 h,TLC监测反应无原料,结束反应。体系冷却至室温,减压蒸馏除去1,4-二氧六环,析出大量白色固体,重新溶于水,1N盐酸调节pH≈6。抽滤得滤饼,并用蒸馏水洗涤,于30 ℃真空干燥8 h,得白色固体纯品化合物(2)[20]15.70 g,产率95.1%。1H NMR(400 MHz,(CD3)2SO):δ12.25(s,1H,31-H),7.73(d,J=8.7 Hz,2H,25,27-H),7.37(d,J=8.2 Hz,2H,12,14-H),7.12(d,J=8.2 Hz,2H,11,15-H),6.90(d,J=8.8 Hz,2H,24,28-H),3.21~3.24(m,4H,19,21-H),2.74(s,2H,10-H),2.25~2.28(m,4H,18,22-H),2.20~2.24(m,2H,2-H),1.99(s,2H,5-H),1.43(t,J=6.1 Hz,2H,1-H),0.97(s,6H,7,8-H)。HR-ESI-MSm/z:439.2147 C26H32N2O2Cl{[M+H]+}。
2结果与讨论
与雅培制药工艺相比[20-21],无需纯化,就可将还原胺化由原来的38.0%提高到90.4%,从而提高了总产率。
与药明康德相比[22],(1)将Suzuki偶联反应放到第二步,不仅克服醛和醇的低沸点且难干燥问题,而且使Suzuki偶联活性增加,重金属催化剂从原来的5.0%当量[24]降低到了2.7‰当量,并且使用常规反应溶剂;(2)从4.6当量三溴化磷[22]改成0.5当量的五氯化磷,总磷量降低了;(3)在卤代反应中用氯代代替了原来的溴代,由于处于烯丙位的氯代物稳定性大于溴代物,降低了烯丙位卤代物的分解副反应使得氯代物收率提高(氯代物94.2%,溴代物86.0%);(4)氯代物与胺反应时不需要NaI或其他活化剂的存在下,就可以直接发生胺烷基化反应;同样由于氯代物的稳定性降低了副反应,胺烷基化产率由溴代物的70.0%[22]提高到了氯代物的96.0%。
3结束语
综上所述,本文合成路线反应条件简单,反应溶剂常规易得,仅需一步纯化,总产率就可达到48.0%,具有操作简单、产率高、反应条件温和等优点。
[1] Lee E F,Clarke O B,Evangelista M,et al.Discovery and molecular characterization of a Bcl-2-regulated cell death pathway in schistosomes[J].Proc Natl Acad Sci USA,2011,108(17):6999-7003.
[2] 李功权.Mcl:1和IAPs家族蛋白介导肝癌细胞抗凋亡作用及其分子机制的研究[D].郑州:郑州第一临床学院,2014.
[3] ADAMS J M,CORY S.The Bcl-2 protein family:Arbiters of cell survival[J].Science,1998,281(5381):1322-1326.
[4] ADAMS J M,CORY S.The Bcl-2 apoptotic switch in cancer development and therapy[J].Oncogene,2007,26(9):1324-1337.
[5] GROSS A,MCDONNELL J M,KORSMEYER S J.BCL-2 family members and the mitochondria in apoptosis[J].Genes & Dev,1999,13(15):1899-1911.
[6] MARTINOU J C,YOULE R J.Mitochondria in Apoptosis:Bcl-2 Family Members and Mitochondrial Dynamics[J].Dev Cell,2011,21(1):92-101.
[7] VANDELFT M F,WEI A H,MASON K D,et al.The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized[J].Cancer Cell,2006,10(5):389-399.
[8] LETAI A.BCL-2:found bound and drugged[J].Trends Mol Med,2005,11(10):442-444.
[9] WANG S,MI(US) S,YANG D,et al.Small molecule antagonists of bcl-2 family proteins[P].US,20030008924,2003-06-09.
[10] WANG G P,NIKOLOVSKA-COLESKA Z,YANG C Y,et al.Structure-based design of potent small-molecule inhibitors of anti-apoptotic Bcl-2 proteins[J].J Med Chem,2006,49(21):6139-6142.
[11] TANG G Z,DING K,NIKOLOVSKA-COLESKA Z,et al.Structure-based design of flavonoid compounds as a new class of small-molecule inhibitors of the anti-apoptotic bcl-2 proteins[J].J Med Chem,2007,50(14):3163-3166.
[12] TANG G Z,YANG C Y,NIKOLOVSKA-COLESKA Z,et al.Pyrogallol-based molecules as potent inhibitors of the antiapoptotic Bcl-2 proteins[J].J Med Chem,2007,50(8):1723-1726.
[13] TANG G Z,NIKOLOVSKA-COLESKA Z,QIU S,et al.Acylpyrogallols as inhibitors of antiapoptotic Bcl-2 proteins[J].J Med Chem,2008,51(4):717-720.
[14] ZHOU H B,CHEN J F,MEAGHER J L,et al.Design of Bcl-2 and Bcl-xL Inhibitors with Subnanomolar Binding Affinities Based upon a New Scaffold[J].J Med Chem,2012,55(10):4664-4682.
[15] BAI L C,CHEN J F,MCEACHERN D,et al.BM-1197:A Novel and Specific Bcl-2/Bcl-xL Inhibitor Inducing Complete and Long-Lasting Tumor Regression In Vivo[J].Plos One,2014,9(6):1-13.
[16] VO D D,GAUTIER F,BARILLE-NION S,etal.Design,synthesis and biological evaluation of new inhibitors of Bax/Bcl-xL interaction in cancer cells[J].Bioorg Med Chem Lett,2014,24(7):1758-1761.
[17] VO D D,GAUTIER F,BARILLE-NION S,et al.Synthesis of new mixed phenol/heterocyclic derivatives and studies of their activity as inhibitors of Bax/Bcl-xL interaction[J].Tetrahedron,2014,70(2):301-311.
[18] LEVOIN N,VO D D,GAUTIER F,et al.A combination of in silico and SAR studies to identify binding hot spots of Bcl-xL inhibitors[J].Bioorg Med Chem,2015,23(8):1747-1757.
[19] BRUNCKO M,OOST T K,BELLI B A,et al.Studies leading to potent,dual inhibitors of bcl-2 and Bcl-xL[J].J Med Chem,2007,50(4):641-662.
[20] PARK C M,BRUNCKO M,ADICKES J,et al.Discovery of an Orally Bioavailable Small Molecule Inhibitor of Prosurvival B-Cell Lymphoma 2 Proteins[J].J Med Chem,2008,51(21):6902-6915.
[21] TS弗兰奇克二世,DR希尔,AR海特,等.用于制备凋亡启动子ABT-263的方法[P].中国专利,102131792,2011-07-20.
[22] 张培权,肖贻崧,张守南,等.4-(4-((2-(4-氯苯基)-5,5-二甲基环己基-1-烯)甲基)哌嗪-1-基)苯甲酸的合成方法[P].中国专利,102584744,2012-07-18.
[23] ZHAI H X,XIONG C.Bcl-2 inhibitors[P].US,2009036035,2009-03-19.
[24] K米勒-莫斯林,B-B托雷,MS维塞尔,等.作为Bcl-2家族蛋白抑制剂用于癌症的治疗的磺酰胺化合物[P].中国专利,102498111,2012-06-13.
[25] P卡萨拉,T勒迪瓜尔赫,O热内斯特,等.新的三元环化合物、它们的制备方法和含有它们的药物组合物[P].中国专利,101270120,2008-09-24.
[26] BAELL J B,LESSENE G L,SLEEBS B E,et al.Arylsulfonamide compounds[P].US,2008061208,2008-05-22.
Synthesis of ABT-263 Intermediate Carboxylic Acid
SHENHuanling,ZHOUGuochun
(School of Pharmaceutical Sciences, Nanjing Tech University, Nanjing 210009, China)
As the key intermediate of an oral Bcl-2 family protein inhibitor ABT-263 (1), the synthetic method for 4-(4-((2-(4-chlorophenyl)-5,5-di-methylcyclohex-1-enyl)methyl) piperazin-1-yl) benzoic acid (2) was reported. It was the reaction of 4,4-dimethylcyclohexanone with phosphorus tribromide andN,N-dimethylformamide to form the first intermediate 2-bromo-5,5-dimethyl-cyclohex-1-ene carbaldehyde (3); and then after the reactions of Suzuki coupling, reduction, halogenation, amidation, hydrolysis and acidification to afford (2). It was prepared in 47% total yield by six steps, significantly higher than the reported yield. The synthesis with simple and mild conditions, convenient workup procedure is suitable for large scale production. The structures of all products were confirmed by1H NMR.
ABT-263; carboxylic acid fragment; synthesis
2016-11-18
国家自然科学基金资助项目(30973621)
申焕玲(1989-),女,河北邯郸人,硕士生,主要从事药物化学方面的研究,(E-mail)shenhuanling@njtech.edu.cn; 周国春(1964-),男,江苏仪征人,博士,博士生导师,主要从事药物化学方面的研究,(E-mail)gczhou@njtech.edu.cn
1673-1549(2017)02-0011-05
10.11863/j.suse.2017.02.03
TQ463+.4
A
