删除Loop区域表面不稳定氨基酸提高 (R)-ω-转氨酶热稳定性
2017-04-16谢东芳吕常江方卉杨卫康胡升赵伟睿黄俊梅乐和
谢东芳,吕常江,方卉,杨卫康,胡升,赵伟睿,黄俊,梅乐和
1 浙江科技学院 生物与化学工程学院,浙江 杭州 310023
2 浙江大学 宁波理工学院 生物与化学工程学院,浙江 宁波 315100
手性胺是一类具有重要价值的医药及精细化工中间体[1]。目前,超过70%的药物,如神经类药物、心血管药物、抗高血压药物、抗感染药物及疫苗等都是以手性胺作为中间体来合成的。例如,抗糖尿病新药Januvia的主要成分西他列汀是R-型胺[2]。化学方法合成手性胺通常成本高、污染大、光学纯度低,而生物合成法具有产物光学纯度高、立体选择性强、反应条件温和、环境友好等优点。目前可以用于手性胺合成的生物催化剂主要有胺脱氢酶[3-5]、人工金属酶[6]、单胺氧化酶[7-8]、亚胺还原酶[9-10]和ω-转氨酶等[2,11]。其中,ω-转氨酶以前手性酮类化合物作为底物,通过立体选择性的转氨基作用实现手性胺的生物合成,且产物纯度高,具有相对更好的工业应用前景[11-12]。
根据 ω-转氨酶作用底物的立体构型,可将ω-转氨酶分为 (S)-ω-TA和 (R)-ω-TA,两者分别作用于 (S)-型底物与 (R)-型底物。相对于 (S)-ω-TA,(R)-ω-TA的研究较少,但其需求量随着手性胺类药物的发展日趋增大[13-14]。显然,挖掘具有潜在应用价值的 (R)-ω-TA对于实现手性胺生物合成和工业生产具有重要价值[2,15-19]。到目前为止,人们已从节杆菌Arthrobacter和河流弧菌Vibrio fluvialis等种属中克隆了多种 (R)-ω-TA基因,通过异源表达和纯化获取了重组 (R)-ω-TA,并研究了相关酶学性质[20-23]。值得关注的是,以来源于土曲霉Aspergilus terreus的 (R)-ω-TA作为生物催化剂用于生产对映体手性胺具有较高的对映体选择性和产率[24]。但该酶热稳定性较差,在实际应用中受到了较大的限制[25]。因此,进一步提高 (R)-ω-TA的热稳定性是拓展该酶应用的有效途径。
近年来,基于温度因子 (B-factor) 策略筛选潜在氨基酸残基的突变位点,并结合蛋白质工程技术实现酶分子的理性改造被认为是提升酶热稳定性的最为有效的手段之一[26-29]。B-factor能反映某个氨基酸残基在整个蛋白质分子的柔性,B-factor值越大,相应部位的构象就越不稳定或柔性越大,不利于酶的稳定性。Reetz等[27]发现对蛋白质结构中 B-factor较高的氨基酸进行饱和突变,部分突变体的热稳定性可以得到明显提高。Cesarini等[30]使用B-factor预测来源于绿脓杆菌Pseudomonas aeruginosa的脂肪酶不稳定的残基位点,并进行饱和突变,与野生酶相比,获得的突变酶在加热孵育后的相对残余活力提高了 7倍。Huang等[25]利用 B-factor并结合能量优化策略对来源于A. terreus的(R)-ω-转氨酶的热稳定性进行改造,在40 ℃条件下,突变酶的半衰期时间比野生酶延长了15.8 min,半失活温度 (T5010) 比野生酶提高了5 ℃。
Loop区域是指暴露在蛋白质结构表面并且具有较高的B-factor的环,理论上这部分区域远离活性中心,对酶的功能没有影响;该区域具有较高的 B-factor,在热运动作用下的形变较大,能显著影响蛋白质结构的稳定性[31]。Thompson和Eisenberg首先提出通过删除Loop区域表面的氨基酸提高蛋白质稳定性的设想[32]。Damnjanović等[31]通过删除来源于链霉菌Streptomyces磷脂酶的D40 Loop区域表面不稳定的氨基酸,成功获得了在70 ℃加热24 h后半衰期比野生酶延长11.7 min的突变酶。
基于以上理论,本文通过 PyMol软件和YASARA软件预测来源于A. terreus的 (R)-ω-TA Loop区域,并分析 Loop区域中各个氨基酸残基的B-factor值,从而确定了该蛋白质结构中不稳定的氨基酸残基位点,再结合定点突变技术对它们进行逐步删除,以期获得热稳定性明显高于野生型的突变体。此外,进一步通过分子动力学及Loop区域分子间相互作用的模拟探讨了热稳定性提高的机制。
1 材料与方法
1.1 材料
1.1.1 酶与试剂
DpnⅠ酶购于 Thermo Scientific公司,PrimeStar Max DNA聚合酶购于 TaKaRa公司(宝生物工程大连有限公司,中国)。引物序列由南京金斯瑞生物科技有限公司合成 (中国)。质粒提取试剂盒、DNA琼脂糖凝胶回收试剂盒、PCR产物纯化试剂盒以及SDS-PAGE凝胶制备试剂盒购于康为世纪生物科技有限公司 (中国)。考马斯亮蓝蛋白质浓度测定试剂盒、Ni-NTA 层析介质、异丙基-β-D-硫代半乳糖苷(IPTG)、卡那霉素、DNA和蛋白质Marker购于生工生物工程 (上海) 股份有限公司。
1.1.2 菌株与质粒
(R)-ω-TA表达宿主菌E. coliBL21(DE3) 及重组质粒 pET-28a-ω-opt-TA为本实验室构建并保藏。
1.1.3 培养基
LB培养基:10 g/L胰蛋白胨,5g/L酵母粉,10 g/L氯化钠,pH 7.0。
1.2 突变文库的构建
根据 (R)-ω-TA 的基因序列 (GenBank Accesion No. XM_001209325) 设计6对定点突变引物,如表 1所示。以 pET-28a-ω-opt-TA质粒为模板,分别采用表 1中的引物,克隆获取目标突变体质粒基因;使用DpnⅠ对PCR反应产物进行消化处理 (37 ℃,2 h),以消除父本模板;采用热击转化法将克隆产物转入E. coliDH 5α感受态细胞中。突变体基因经通用生物系统 (安徽) 有限公司测序验证后进一步转化至E. coliBL21(DE3) 感受态细胞中,获取目标重组菌株。
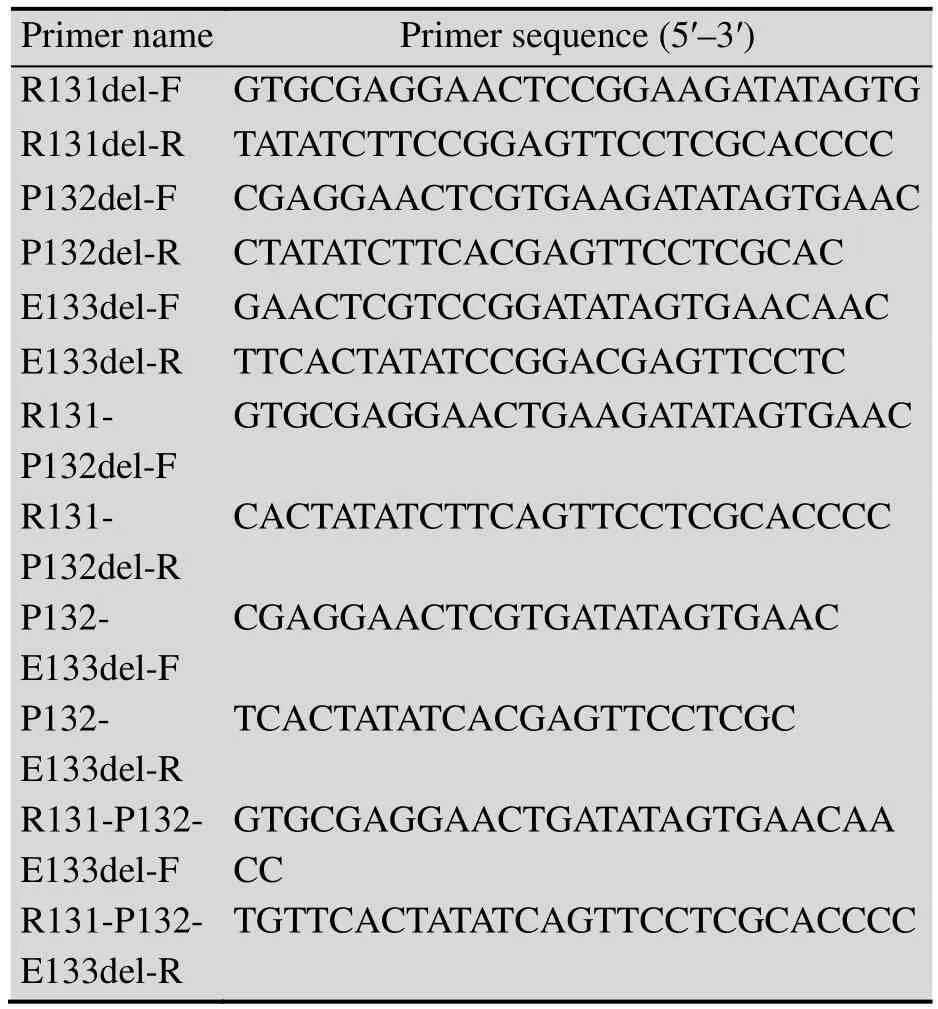
表1 定点突变引物及其序列Table 1 Primers used for site-directed mutagenesis
1.3 野生酶和突变酶的表达和纯化
挑取野生型及突变体单菌落接种至 5 mL含50 μg/mL卡那霉素的LB培养基中,37 ℃、200 r/min条件下培养过夜。随后,将菌液以1%的接种量 (V/V) 转接至200 mL含50 μg/mL卡那霉素的 LB培养基中,37 ℃、180 r/min培养至OD600值为0.4−0.6时加入终浓度为0.5 mmol/L的IPTG,并于25 ℃、150 r/min条件下诱导培养18 h后收集菌体细胞。
将收获的菌体细胞经磷酸盐缓冲液(50 mmol/L,pH 8.0) 离心洗涤 2次后重悬于20 mL破胞缓冲液中 (50 mmol/L磷酸二氢钠,300 mmol/L氯化钠,20 mmol/L咪唑,pH 8.0),超声处理破碎细胞 (超声条件:功率 300 W,工作3 s,间歇6 s,超声15 min),离心30 min(12000 r/min,4 ℃),收集上清液,即得到含有(R)-ω-TA的粗酶液。粗酶液经 0.45 μm滤膜过滤后采用Ni-NTA亲和层析获取目标重组蛋白,并分别采用 SDS-PAGE电泳和考马斯亮蓝法测定纯化后的蛋白纯度及浓度。
1.4 野生酶和突变酶的热稳定性测定
分别将纯化后的野生酶和突变酶在25−55 ℃水浴中孵育10 min,孵育结束后迅速放置在冰上冷却 10 min。底物溶液用磷酸盐缓冲液 (50 mmol/L,pH 8.0) 配制,200 μL 的反应体系中包括180 μL的底物溶液 (0.25% DMSO、2.5 mmol/L (R)-(+)-α-甲基苄胺 ((R)-α-MBA)、2.5 mmol/L 丙酮酸以及 0.1 mmol/L PLP),20 μL纯酶液 (约0.3 mg/mL),采用MD190酶标仪检测波长在245 nm处相应的酶活力,测定方法具体参照文献[33]。以温度为横坐标,以热处理后与处理前酶活力的比值为纵坐标作图,采用 Origin 8.0软件进行Boltzmann S型函数拟合,计算半失活温度(T5010)。
将野生酶和突变酶在 40 ℃下分别孵育2−50 min,孵育结束后迅速放置在冰上冷却10 min,采用上述方法测定酶活力。以时间为横坐标,以热处理后与处理前比活力的比值为纵坐标作图,通过Origin 8.0软件拟合非线性方程y=exp (–kd·t),一阶速率常数 (kd) 经非线性回归确定,并计算酶活力降低为 50%时所对应的半衰期(t1/2)。
1.5 野生酶和突变酶的最适反应温度测定
在180 μL预热的底物溶液中加入20 μL纯酶液(约 0.3 mg/mL),并迅速置于不同温度 (25−60 ℃)的恒温混匀仪中,400 r/min条件下反应3 min。随后,将含有上述混合物的离心管迅速放入100 ℃的水浴锅中煮沸10 min;冰上放置10 min后利用酶标仪检测波长245 nm处的OD值。
1.6 野生酶和突变酶的动力学参数测定
采用50 mmol/L的磷酸缓冲液 (pH 8.0) 配制不同浓度 (0−3 mmol/L) 的底物 (R)-α-MBA 和丙酮酸溶液。使用MD190酶标仪测定在不同底物浓度条件下的酶活力,以底物浓度[S]对反应速率[V]作图,通过非线性拟合计算相应的Km和Vmax值,然后根据kcat=Vmax/[E0] ([E0]为酶初始浓度,单位mmol/L) 计算求得kcat和催化效率kcat/Km。
1.7 野生酶和突变酶的分子动力学模拟
在Protein Data Bank (PDB数据库) 中搜索来源于A. terreus的 (R)-ω-转氨酶晶体结构,选择分辨率为1.6 Å的结构 (PDB ID: 4CE5)[24]作为模板,采用 Swiss Model建模方法 (http://swissmodel.expasy.org) 构建突变体的三维结构,并采用YASARA软件中的Amber14力场,对野生酶和突变酶在313 K和400 K温度下进行了时长为10 ns的动力学模拟。模拟过程为:以PDB格式的野生型和突变体 (R)-ω-TA的三维结构作为分子动力学模拟的初始结构,经加氢处理后将晶体结构放置于 10 Å×10 Å×10 Å 的立方体盒子中,并以 0.98 g/L的密度填充水分子,并向体系中添加合适的抗衡离子 (Na+/Cl–),使模拟系统呈电中性。范德华力相互作用的短程截断距离为7.86 Å,采用 Particle Mesh Ewald方法计算长程静电相互作用。
2 结果与分析
2.1 (R)-ω-转氨酶的Loop区域预测及分析
B-factor反映了氨基酸残基在整个蛋白质分子中的柔性,B-factor值越大,相应部位的构象柔性就越大。本研究利用 PyMOL软件和YASARA软件预测了Loop区域的B-factor,结果如图 1所示。除了N-端和C-端以外,Loop区域表面的氨基酸残基具有较高的B-factor,其中Arg131、Pro132和Glu133处于Loop区域的黄色颜色部分,是B-factor最大的位点。与(R)-ω-TA中所有原子的平均B-factor (21) 相比,131位点的精氨酸 (Arg131)、132位点的脯氨酸 (Pro132)及133位点的谷氨酸 (Glu133) 的B-factor分别高出41、23与31。因此,选取这3个关键氨基酸残基用于考察其缺失对于 (R)-ω-TA热稳定性的影响。
2.2 野生酶和突变酶的表达和纯化
对 (R)-ω-TA的Loop区域 (图1) 中上述3个不稳定的氨基酸残基进行了逐一删除,获取了相应的重组突变体酶,并分别命名为R131del、P132del和E133del。此外,同时删除相邻的2个氨基酸残基及同时删除这3个氨基酸残基的重组突变体也被进一步构建并提取纯酶,分别命名为R131-P132del、P132-E133del和R131-P132-E133del。经 Ni-NTA亲和层析纯化后的野生酶及突变酶的SDS-PAGE检测结果如图2所示,纯化后的野生酶和突变酶的电泳条带清晰且单一,其分子量约为36 kDa,与理论分子量相一致,为后续实验的进行奠定了基础。
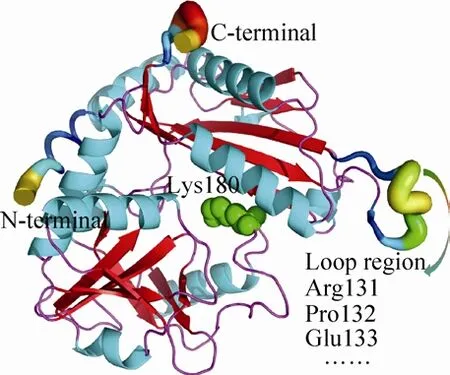
图1 (R)-ω-转氨酶的三维结构Fig. 1 Simulated three-dimensional structure of(R)-ω-TA. The most flexible region according to B-factors from x-ray crystal structure is used as the target for of residue deletion. The active center (Lys 180)is shown as green spheres.
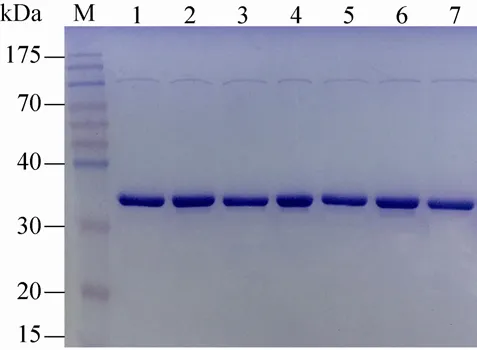
图2 野生酶和突变酶的SDS-PAGE分析Fig. 2 SDS-PAGE analysis of wide type enzyme and mutant enzymes. M: protein marker; lane 1: wide type enzyme; lane 2: mutant enzyme R131del; lane 3: mutant enzyme P132del; lane 4: mutant enzyme E133del; lane 5:mutant enzyme R131-P132 del; lane 6: mutant enzyme P132-E133del; lane7: mutant enzyme R131-P132-E133del.
2.3 野生酶和突变酶的热稳定性
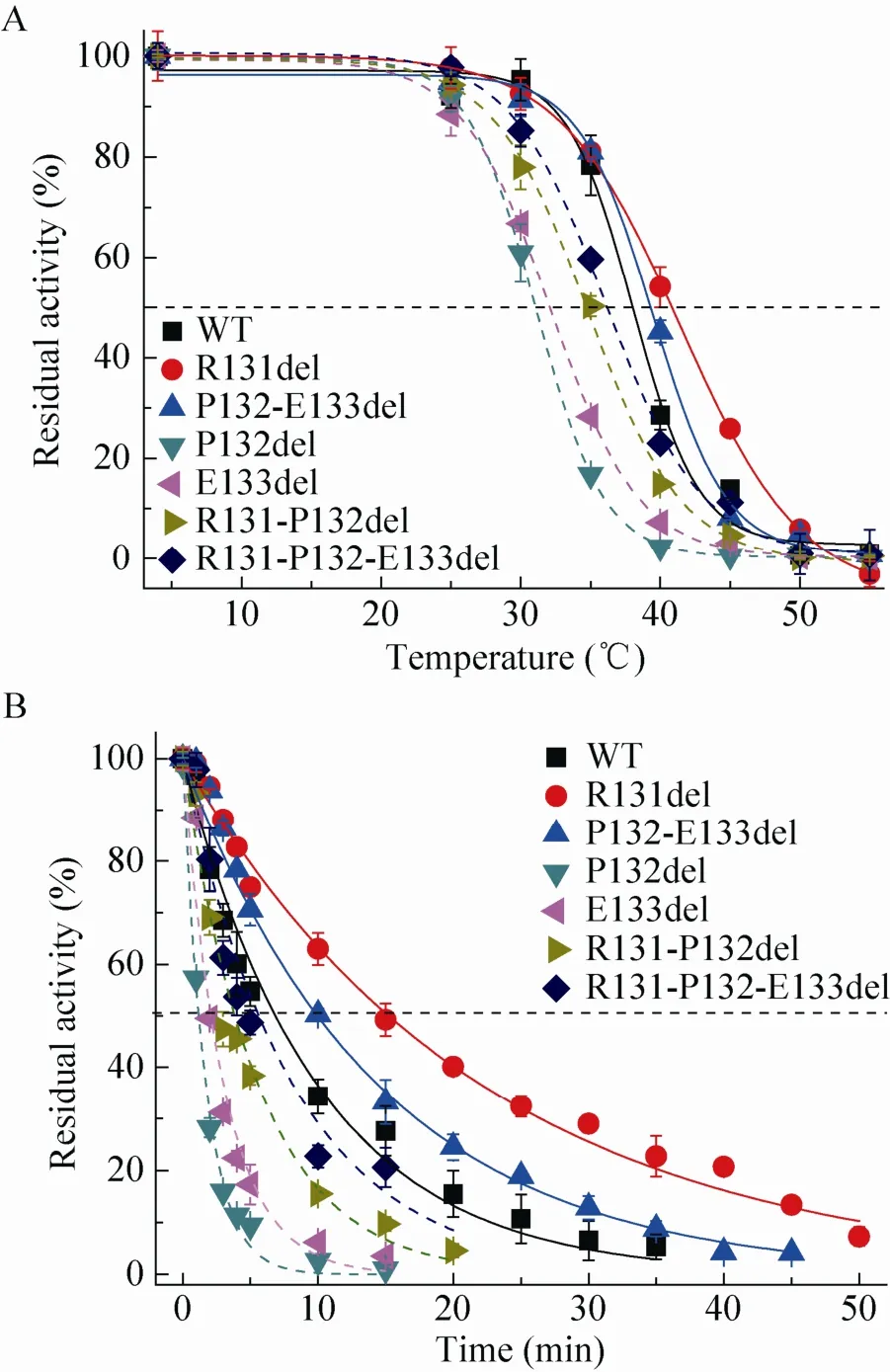
图3 野生酶和突变酶的热稳定性Fig. 3 Thermostability analysis of wide type enzyme and mutant enzymes. (A) Thermal inactivation of wild type enzyme and mutant enzymes at different temperatures over 10 min (T5010); (B) Thermal inactivation half-life (t1/2) of wild type enzyme and mutant enzymes at 40 ℃. The error bars show standard deviation calculated for three replicate experiments.
按照 1.4所述的方法测定了野生酶与突变酶的半失活温度 (T5010) 和半衰期 (t1/2),结果如图 3所示。由图 3可知,突变酶 R131del(41.1 ℃) 和突变酶P132-E133del (39.4 ℃) 的T5010比野生酶 (38.5 ℃) 提高了 2.6 ℃和0.9 ℃,在 40 ℃下的t1/2分别为 15.0 min和10.0 min,为野生酶 (6.9 min) 的2.2倍和1.5倍;而突变酶 P132del (31.1 ℃)、突变酶 E133del(32.1 ℃)、突变酶 R131-P132del (34.9 ℃) 和突变酶 R131-P132del-E133del (36.6 ℃) 的T5010却比野生酶降低了 7.4 ℃、6.4 ℃、3.6 ℃和1.9 ℃,在 40 ℃下的t1/2也比野生型减少了5.7、4.8、1.7和 0.8 min。由此表明,突变酶R131del较野生酶的热稳定性有了较大的提高;突变酶P132-E133del较野生酶的热稳定性稍有提升;然而突变酶P132del、E133del、R131-P132del和 R131-P132-E133del的热稳定性比野生酶有所降低。它们的热稳定性顺序分别为:突变酶R131del>突变酶 P132-E133del>野生酶>突变酶R131-P132del-E133del>突变酶 R131-P132del>突变酶 E133del>突变酶 P132del。基于以上结果,选取突变酶R131del与突变酶P132-E133del进行后续的实验研究。
2.4 野生酶和突变酶最适温度测定
按照1.5所述的方法测定了野生酶与突变酶的最适反应温度,结果如图4所示。由图4可知,野生酶、突变酶R131del和突变酶P132-E133del的最适反应温度均为 45 ℃。但是当温度超过45 ℃时,突变酶R131del和突变酶P132-E133del的相对活力显著高于野生酶并且在 60 ℃时的相对活力仍保持在 50%以上,而野生型酶在60 ℃的相对活力仅有30%。
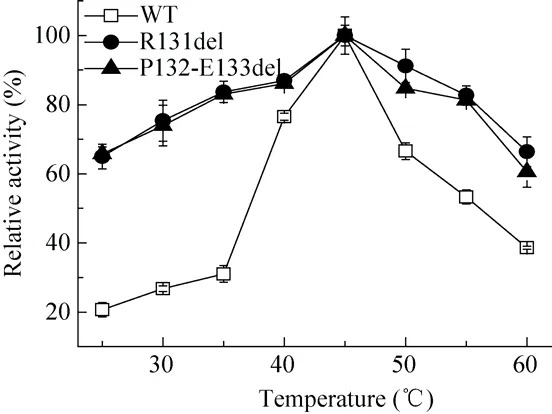
图4 野生酶和突变酶的最适温度Fig. 4 The optimum catalytic temperature analysis of wide type enzyme and mutant enzymes. The error bars show standard deviation calculated for three replicate experiments.
2.5 野生酶和突变酶的动力学参数
分别以 (R)-α-MBA和丙酮酸为底物,按照1.6所述的方法测定野生酶、突变酶R131del和突变酶 P132-E133del的米氏动力学常数,采用Hyperbola函数进行非线性拟合得到动力学参数,结果见表 2。由表 2可知,野生酶对丙酮酸的催化效率 (kcat/Kmpyruvate) 为 2.22 L/(mmol·s),突变酶R131del和P132-E133del的kcat/Kmpyruvate分别为野生酶的 1.05倍和 0.91倍;野生酶对 (R)-α-MBA的催化效率 (kcat/Kmα-MBA) 为 2.82 L/(mmol·s),突变酶 R131del和 P132-E133del的kcat/Kmα-MBA分别为野生酶的0.97和1.39 倍。由此表明,突变酶 R131del和 P132-E133del热稳定性得到提高的同时,并未显著降低酶的催化效率。

表2 野生酶和突变酶酶学参数比较Table 2 Kinetic analysis of wide type and mutant enzyme
2.6 野生酶和突变酶的分子动力学模拟
分子动力学模拟是一种利用计算机模拟蛋白质结构中原子的物理运动的方法,通过模拟的计算机结果可以计算骨架原子的root mean square fluctuation (RMSF) 值,从而确定蛋白质的热敏感或构象柔性区域[33]。RMSF值反映了蛋白质结构中单个氨基酸残基的稳定性,并且它与蛋白质的热稳定性呈负相关[26]。将野生酶和突变酶在313 K和400 K温度下分别进行了时长为10 ns的动力学模拟,获得的RMSF 值如图5所示。由图5A和 5C可知,在 313K温度下,野生酶、突变酶R131del和突变酶P132-E133del之间没有明显差异。
由图 5B可知,在 400 K温度下突变酶R131del所有氨基酸残基的RMSF值均低于野生酶,这表明突变酶R131del在高温下具有更稳定的结构,从而导致它的热稳定性高于野生酶。由图5D可知,在400 K温度下突变酶P132-E133del在Loop区域的RMSF值与野生酶差异不显著。
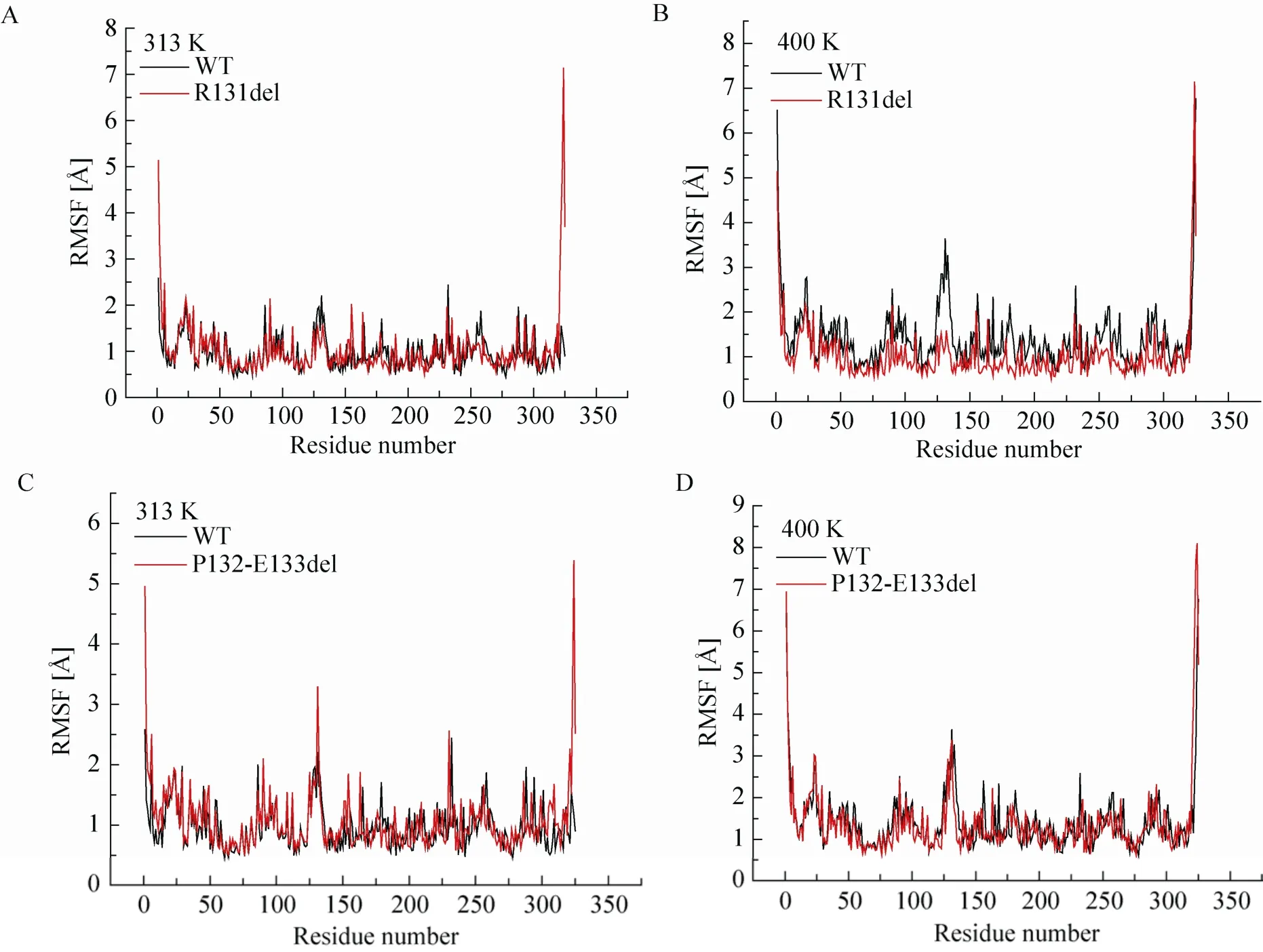
图5 野生型酶和突变体酶的MD模拟Fig. 5 Molecular dynamics (MD) simulation analysis of wild-type and mutant enzymes for 10 ns at 313 K and 400 K using YASARA. (A) RMSF values for mutant enzyme R131del at 313 K during a 10 ns simulation. (B) RMSF values for mutant enzyme R131del at 400 K during a 10 ns simulation. (C) RMSF values for mutant enzyme P132-E133del at 313 K during a 10 ns simulation. (D) RMSF values for mutant enzyme P132-E133del at 400 K during a 10 ns simulation.
3 讨论
目前,研究者已经通过化学修饰、易错PCR、定点突变、蛋白质融合、DNA改组等手段有效地提高了多种酶的热稳定性[34]。基于B-factor值确定影响酶热稳定性的关键氨基酸残基,进而利用蛋白质工程技术对酶分子进行理性改造可以有效地提高其热稳定性[26-29]。本文利用此方法成功筛选到了正突变酶 R131del和 P132-E133del。
实验结果表明,突变酶 (R131del) 和突变酶 (P132-E133del) 半失活温度分别为 41.1 ℃和39.4 ℃,比野生酶提高了2.6 ℃和0.9 ℃,在40 ℃下的半衰期分别为15.0 min和10.0 min,分别是野生酶的2.2倍和1.5倍。采用YASARA软件对野生酶和突变酶 (R131del、P132-E133del)进行分子动力学模拟。研究显示,在400 K和10 ns的模拟条件下,突变酶 R131del的 RMSF值比野生型低,这表明突变酶 R131del不稳定的Loop区域柔性降低,致使突变酶具有更高的热稳定性。通过在线服务器 (http://pic.mbu.iisc.ernet.in/)进一步对野生酶和突变酶 (R 131 d e l、P132-E133del) 的 Loop区域进行了分子间相互作用的模拟分析,探讨突变酶稳定性提高的机制,选取疏水相互 (距离设为5 Å) 和氢键选项,具体分析方法参照参考文献[35],模拟结果如图6所示。由图6A可知,野生酶的131位点是精氨酸,并不能与其他相邻的残基形成疏水相互作用,但当 131位点精氨酸被删除后,突变酶R131del的 131位点的脯氨酸 (Pro131) 与 134位点的异亮氨酸 (Ile134) 之间增加了疏水相互作用,且两者的空间距离为 4.4 Å。Pace等[36]研究了疏水相互作用和氢键对22个分子量大小不同的蛋白质 (氨基酸残基数量在36–534之间)稳定性的影响,研究表明疏水相互作用对这 22个蛋白质的稳定性平均贡献为60%±4%;氢键对这 22个蛋白质的稳定性平均贡献为 40%±4%。因此,当 131位点精氨酸被删除后,突变酶R131del在Loop区域增加了相互疏水作用,从而使其稳定性得到了提高。由图6B可知,与野生型酶相比,突变酶P131-E133del (氨基酸数量为323个) 的Loop区域内主要增加了4个的氢键。它们分别是天冬酰胺 (Asn135) 上的 OD1原子与侧链的精氨酸 (Arg90) 上的N原子 (空间距离为1.9 Å) 和CB原子 (空间距离为3.6 Å)形成了 2个氢键;天冬酰胺 (Asn135) 的 ND2原子与精氨酸 (Arg122) 上的 N 原子 (空间距离分别为2.0 Å和3.6 Å) 形成了2个氢键。因此,Loop区域增加的分子间氢键使得突变酶P131-E133del的热稳定性得到了提高。

图 6 突变酶 R131del (A) 和突变酶 P132-E133del(B) Loop区域的分子间相互作用模拟Fig. 6 Modeling analysis of intermolecular interactions in loop region of mutant enzymes R13del and P132-E133del. (A) In the mutant enzyme R131del, Ile 134 was involved in hydrophobic interaction with the neighbouring hydrophobic amino acid Pro131. (B) In the mutant enzyme P132-E133del, Asn135 was involved in four hydrogen bonds interaction with the neighbouring amino acids Arg90 and Arg122.
本文针对来源于A. terreus的 (R)-ω-TA热稳定性差的缺陷,基于“木桶理论”,对蛋白质柔性区域的“短板”进行刚性改造。通过删除 Loop区域表面不稳定氨基酸的方法,从构建的 6个突变体中成功筛选到热稳定性提高的突变酶R131del和P132-E133del,并利用分子动力学模拟和分子间相互作用的模拟,初步揭示了(R)-ω-TA热稳定性提高的机制,这为其他酶的热稳定性改造提供了方法学指导。
[1] Breuer M, Ditrich K, Habicher T, et al. Industrial methods for the production of optically active intermediates. Angew Chem, 2004, 43(7): 788–824.
[2] Savile CK, Janey JM, Mundorff EC, et al.Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture.Science, 2010, 329(5989): 305–309.
[3] Ye LJ, Toh HH, Yang Y, et al. Engineering of amine dehydrogenase for asymmetric reductive amination of ketone by evolvingRhodococcusphenylalanine dehydrogenase. ACS Catal, 2015,5(2): 1119–1122.
[4] Zheng GW, Xu JH. New opportunities for biocatalysis: driving the synthesis of chiral chemicals. Curr Opin Biotechnol, 2011, 22(6):784–792.
[5] Abrahamson MJ, Wong JW, Bommarius AS. The evolution of an amine dehydrogenase biocatalyst for the asymmetric production of chiral amines.Adv Synth Catal, 2013, 355(9): 1780–1786.
[6] Ringenberg MR, Ward TR. Merging the best of two worlds: artificial metalloenzymes for enantioselective catalysis. Chem Commun, 2011, 47(30): 8470–8476.
[7] Li GY, Ren J, Yao PY, et al. Deracemization of 2-methyl-1,2,3,4-tetrahydroquinoline using mutant cyclohexylamine oxidase obtained by iterative saturation mutagenesis. ACS Catal, 2014, 4(3):903–908.
[8] Höhne M, Bornscheuer UT. Biocatalytic routes to optically active amines. Chem Cat Chem, 2009,1(1): 42–51.
[9] Leipold F, Hussain S, Ghislieri D, et al.Asymmetric reduction of cyclic imines catalyzed by a whole-cell biocatalyst containing an (S)-Imine reductase. Chem Cat Chem, 2013, 5(12): 3505–3508.
[10] Li H, Tian P, Xu JH, et al. Identification of an imine reductase for asymmetric reduction of bulky dihydroisoquinolines. Org Lett, 2017, 19(12):3151–3154.
[11] Pavlidis IV, Weiß MS, Genz M, et al. Identification of (S)-selective transaminases for the asymmetric synthesis of bulky chiral amines. Nat Chem, 2016,8(11): 1076–1082.
[12] Hanson RL, Davis BL, Chen YJ, et al. Preparation of (R)-amines from racemic amines with an(S)-amine transaminase fromBacillus megaterium.Adv Synth Catal, 2008, 350(9): 1367–1375.
[13] Mathew S, Yun H. ω-Transaminases for the production of optically pure amines and unnatural amino acids. Org Catal, 2012, 2(6): 993–1001.
[14] Jiang JJ, Chen X, Feng JH, et al. Substrate profile of an ω-transaminase fromBurkholderia vietnamiensisand its potential for the production of optically pure amines and unnatural amino acids. J Mol Catal B:Enzym, 2014, 100: 32–39.
[15] Midelfort KS, Kumar R, Han S, et al. Redesigning and characterizing the substrate specificity and activity ofVibrio fluvialisaminotransferase for the synthesis of imagabalin. Protein Eng, Des Sel,2013, 26(1): 25–33.
[16] Mutti FG, Fuchs CS, Pressnitz D, et al. Amination of ketones by employing two new (S)-selective ω-transaminases and the his-tagged ω-TA fromVibrio fluvialis. Eur J Org Chem, 2012, 2012(5):1003–1007.
[17] Park E, Kim M, Shin JS. One-pot conversion of L-threonine into L-homoalanine: biocatalytic production of an unnatural amino acid from a natural one. Adv Synth Catal, 2010, 352(18): 3391–3398.
[18] Shin JS, Kim BG. Exploring the active site of amine: pyruvate aminotransferase on the basis of the substrate structure-reactivity relationship: how the enzyme controls substrate specificity and stereoselectivity. J Org Chem, 2002, 67(9):2848–2853.
[19] Yun H, Cho BK, Kim BG. Kinetic resolution of(R,S)-sec-butylamine using omega-transaminase fromVibrio fluvialisJS17 under reduced pressure.Biotechnol Bioeng, 2004, 87(6): 772–778.
[20] Höhne M, Schätzle S, Jochens H, et al. Rational assignment of key motifs for function guidesin silicoenzyme identification. Nat Chem Biol, 2010,6(11): 807–813.
[21] Iwasaki A, Matsumoto K, Hasegawa J, et al. A novel transaminase, (R)-amine: pyruvate aminotransferase, fromArthrobactersp. KNK168(FERM BP-5228): purification, characterization,and gene cloning. Appl Microbiol Biotechnol,2012, 93(4): 1563–1573.
[22] Mutti FG, Fuchs CS, Pressnitz D, et al.Stereoselectivity of four (R)-selective transaminases for the asymmetric amination of ketones. Adv Synth Catal, 2011, 353(17): 3227–3233.
[23] Schätzle S, Steffen-Munsberg F, Thontowi A, et al.Enzymatic asymmetric synthesis of enantiomerically pure aliphatic, aromatic and arylaliphatic amines with (R)-selective amine transaminases. Adv Synth Catal, 2011, 353(13): 2439–2445.
[24] Łyskowski A, Gruber C, Steinkellner G, et al.Crystal structure of an (R)-selective ω-transaminase fromAspergillus terreus. PLoS ONE, 2014, 9(1):e87350.
[25] Huang J, Xie DF, Feng Y. Engineering thermostable (R)-selective amine transaminase fromAspergillus terreusthroughinsilicodesign employing B-factor and folding free energy calculations. Biochem Biophys Res Commun,2017, 483(1): 397–402.
[26] Huang J, Jones BJ, Kazlauskas RJ. Stabilization of an α/β-hydrolase by introducing proline residues:salicylic acid binding protein 2 fromTobacco.Biochemistry, 2015, 54(28): 4330–4341.
[27] Reetz MT, Carballeira JD, Vogel A. Iterative saturation mutagenesis on the basis of B factors as a strategy for increasing protein thermostability.Angew Chem, 2006, 45(46): 7745–7751.
[28] Xie Y, An J, Yang GY, et al. Enhanced enzyme kinetic stability by increasing rigidity within the active site. J Biol Chem, 2014, 289(11): 7994–8006.
[29] Parthasarathy S, Murthy MRN. Protein thermal stability: insights from atomic displacement parameters(Bvalues). Protein Eng Des Sel, 2000, 13(1): 9–13.
[30] Cesarini S, Bofill C, Pastor FIJ, et al. A thermostable variant ofP. aeruginosacold-adapted LipC obtained by rational design and saturation mutagenesis. Process Biochem, 2012, 47(12):2064–2071.
[31] Damnjanović J, Nakano H, Iwasaki Y. Deletion of a dynamic surface loop improves stability and changes kinetic behavior of phosphatidylinositol-synthesizingStreptomycesphospholipase D. Biotechnol Bioeng,2014, 111(4): 674–682.
[32] Thompson MJ, Eisenberg D. Transproteomic evidence of a loop-deletion mechanism for enhancing protein thermostability. J Mol Biol,1999, 290(2): 595–604.
[33] Purmonen M, Valjakka J, Takkinen K, et al.Molecular dynamics studies on the thermostability of family 11 xylanases. Protein Eng Des Sel, 2007,20(11): 551–559.
[34] Zhang SP, Liu ZQ, Zheng YG. Research progress of modification forCandida antarcticaLipase B.Genomics Appl Biol, 2011, 30(2): 243–250 (in Chinese).章素平, 柳志强, 郑裕国. 南极假丝酵母脂肪酶B的修饰与改造. 基因组学与应用生物学, 2011,30(2): 243–250.
[35] Tina KG, Bhadra R, Srinivasan N. PIC: protein interactions calculator. Nucleic Acids Res, 2007,35: W473–W476.
[36] Pace CN, Fu HL, Fryar KL, et al. Contribution of hydrophobic interactions to protein stability. J Mol Biol, 2011, 408(3): 514–528.
