重组葡萄糖脱氢酶的表达及辅酶再生应用研究
2017-04-13李凌凌刘曜宁吕早生杨忠华左振宇宋采薇
李凌凌,刘曜宁,吕早生,杨忠华,左振宇,宋采薇
(武汉科技大学化学与化工学院,湖北 武汉,430081)
重组葡萄糖脱氢酶的表达及辅酶再生应用研究
李凌凌,刘曜宁,吕早生,杨忠华,左振宇,宋采薇
(武汉科技大学化学与化工学院,湖北 武汉,430081)
为解决异源表达酮还原酶域EryKR1的重组大肠杆菌EscherichiacoliBL21(pET28a-eryKR1)催化环己酮还原时消耗的氢供体NADPH再生的问题,构建了克隆枯草芽孢杆菌葡萄糖脱氢酶基因gdh的重组菌E.coliBL21(pET28a-gdh),其中的gdh基因经Nucleotide BLAST功能分析显示与枯草芽孢杆菌9902的gdh基因序列(登录号为EF626962.1)的一致性达到100%。SDS-PAGE检测显示该重组菌经IPTG诱导后可以高效表达出葡萄糖脱氢酶(GDH),其表达量占全菌可溶性蛋白质的64%。GDH粗酶液的比酶活为137.90 U/mg。通过气相色谱检测添加了E.coliBL21(pET28a-gdh)的E.coliBL21(pET28a-eryKR1)环己酮还原反应体系中的环己醇含量,结果显示加入重组GDH的双重组菌耦合反应体系中环己醇的产率为82.21%,是未添加GDH的反应体系对应值的3.23倍,表明重组GDH可以为EryKR1还原环己酮系统解决辅酶再生问题。
葡萄糖脱氢酶;重组菌;异源表达;辅酶再生;生物催化;环己酮;枯草芽孢杆菌
利用氧化还原酶催化潜手性羰基化合物不对称还原合成手性醇是生物技术中一个具有巨大发展前景的研究领域。但是,这个催化反应需要大量价格昂贵且稳定性低的辅酶协助才能进行,从而极大地阻碍了氧化还原酶的大规模应用,因此构建低成本、高效的辅酶再生系统是解决问题的关键[1]。解决辅酶再生的方法有化学法、电化学法、光化学法和酶法[2],其中酶法更受青睐。酶法辅酶再生中负责将NAD(P)+转化成NAD(P)H的酶有葡萄糖脱氢酶(GDH)和甲酸脱氢酶等。葡萄糖脱氢酶具有高稳定性和强特异性,而且能够以廉价的葡萄糖为底物进行反应,因此有着广泛的应用前景。目前,研究人员已经在巨大芽孢杆菌、枯草芽孢杆菌、甲基营养菌、弱氧化醋酸杆菌、嗜酸热原体等微生物中分离到了葡萄糖脱氢酶[3-6],其中枯草芽孢杆菌葡萄糖脱氢酶的产量高,纯化方便,已被广泛应用于辅酶的再生。酶法辅酶再生包括游离酶或固定化酶催化以及全细胞催化等方式[2]。游离酶或固定化酶催化具有可控性强、可连续操作且易于放大的特点,但其成本过高,酶的稳定性和寿命也有限;而全细胞催化省去了复杂的分离和纯化步骤,操作比较方便。
克隆了糖多孢红霉菌聚酮合成酶模块1中酮还原酶域基因的重组大肠杆菌EscherichiacoliBL21(pET28a-eryKR1)可以催化潜手性脂环酮的不对称还原[7],但此反应需要消耗大量昂贵的辅酶NADPH。本研究旨在构建重组表达枯草芽孢杆菌葡萄糖脱氢酶的大肠杆菌,通过基因工程手段实现葡萄糖脱氢酶的异源高效表达,并采用双重组菌耦合的全细胞催化方法,解决酮还原酶域催化的还原反应中的辅酶再生问题(其中以环己酮替代潜手性脂环酮作为底物),以期降低生产成本,提高市场价值。
1 实验材料与方法
1.1 材料
1.1.1 菌种、质粒和培养基
枯草芽孢杆菌(Bacillussubtilis168)、大肠杆菌表达载体pET28a质粒及重组菌E.coliBL21(pET28a-eryKR1)为本实验室保藏;大肠杆菌BL21(E.coliBL21(DE3))为本实验宿主菌,购自康为世纪生物科技有限公司。
LB培养基按照文献[8]配制,用于培养大肠杆菌BL21和枯草芽孢杆菌。筛选抗性菌株时,加入终浓度为100 μg/mL的卡拉霉素。
1.1.2 主要试剂
Lysozyme、蛋白酶K、pfu DNA聚合酶、限制性内切酶NdeⅠ和EcoRⅠ、T4DNA连接酶、DNA Ladder Marker、Protein Marker、DNA Loading Buffer、微量琼脂糖凝胶DNA回收试剂盒及高纯度质粒小提试剂盒均为康为世纪生物科技有限公司产品。
1.2 实验方法
1.2.1 枯草芽孢杆菌葡萄糖脱氢酶基因的PCR扩增
根据GenBank中登录号为EF626962.1的枯草芽孢杆菌9902的葡萄糖脱氢酶基因序列设计合成引物。为了便于葡萄糖脱氢酶基因的克隆,在正向引物和反向引物前分别加上NdeⅠ和EcoRⅠ酶切位点,并加上适量保护碱基,设计的引物如下:Gdh1X:5’-gccatatgTATCCGGATTTAAAAGGAAAAGTCGTCGC-3’(gc为保护性碱基,catatg为NdeⅠ酶切位点);Gdh2:5’-ggaattcTTAACCGCGGCCTGCCTGGAA-3’(g为保护性碱基,gaattc为EcoRⅠ酶切位点)。引物提交给武汉擎科创新生物科技有限公司合成。
PCR扩增体系:CTAB法提取的枯草芽孢杆菌基因组DNA 2 μL,dNTP(each 2.5 mmol/L)2.5 μL,10×pfu DNA聚合酶缓冲液2.5 μL,DMSO 2.5 μL,引物Gdh1X(25 μmol/L)0.5 μL,引物Gdh2(25 μmol/L)0.5 μL,pfu DNA聚合酶(5 U/μL)0.5 μL,最后补无菌水至25 μL;PCR扩增程序参照文献[7]。
1.2.2 枯草芽孢杆菌葡萄糖脱氢酶基因的克隆
按照微量琼脂糖凝胶DNA回收试剂盒的说明书回收纯化PCR产物中的目标片段、PCR产物和质粒的双酶切产物。按照高纯度质粒小提试剂盒的说明书提取质粒。PCR扩增产物及载体的双酶切、连接方法和连接产物的转化方法按照文献[9]进行。重组质粒由武汉擎科创新生物科技有限公司测序。
1.2.3 重组葡萄糖脱氢酶的诱导表达
按照5%的接种量将构建的重组菌种子液接种到50 mL含100 μg/mL 卡拉霉素的LB液体培养基中,37 ℃、150 r/min培养3 h左右加入IPTG(终浓度为1 mmol/L),30 ℃、150 r/min诱导6 h。离心(4 ℃,12 000g,10 min)收集菌体沉淀,用0.1 mol/L的磷酸钾缓冲液(pH 7.0)洗涤重悬后,超声波破碎(400 W,工作3 s间歇5 s,10 min)。离心(4 ℃,12 000g,10 min)收集上清液(即粗酶液),测定其酶活、蛋白质含量并进行SDS-PAGE电泳分析。
测定酶活的反应体系(3 mL):粗酶液20 μL、20 mmol/L NADP+300 μL、125 mmol/L葡萄糖2.4 mL和0.1 mol/L磷酸钾缓冲液(pH 7.0)280 μL。该体系置于37 ℃反应30 min,期间在340 nm下测量NADPH的吸光值变化曲线,根据曲线斜率计算单位时间内的NADPH含量变化情况。酶活单位U定义为在本实验条件下,1 min催化还原1 μmol NADP+生成NADPH所需要的酶量。以上测定过程重复3次。采用Bradford法测定蛋白质含量,以牛血清白蛋白(BSA)为标准品[8];根据文献[8]进行SDS-PAGE分析;
1.2.4 双重组菌耦合发酵检测辅酶再生系统能力
双重组菌耦合的发酵体系(20 mL):0.4 gE.coliBL21(pET28a-eryKR1),0.1 gE.coliBL21(pET28a-gdh),2.5 mg/mL 葡萄糖、0.2 mmol/L NADPH、16.6 μmol/mL 环己酮和0.1 mol/L磷酸钾缓冲液(pH 6.0)。该体系置于30 ℃、100 r/min振荡反应6 h。反应液离心(4 ℃,12 000g,10 min)收集上清液并用等体积乙酸乙酯萃取,按照文献[7]的气相色谱条件检测有机相组分。
2 结果与分析
2.1 pET-gdh表达载体的构建
以提取的枯草芽孢杆菌基因组DNA为模板、以Gdh1X和Gdh2的合成引物进行PCR扩增,产物的1.0% 琼脂糖凝胶电泳检测结果如图1所示。由图1可见,在750 bp附近存在目的片段,这与目标基因gdh的长度786 bp基本一致。对质粒pET28a和gdh基因的PCR回收产物进行双酶切,回收后再进行连接反应。连接产物转化到大肠杆菌BL21中,挑选出疑似阳性克隆,从中提取出质粒进行双酶切鉴定,如图2所示。质粒经双酶切后,将其中连接长度约为750 bp的gdh基因片段切下来,证实了疑似阳性克隆中提取出来的质粒就是已克隆了枯草芽孢杆菌葡萄糖脱氢酶基因的重组质粒pET28a-gdh,命名为E.coliBL21(pET28a-gdh)。
对重组质粒pET28a-gdh进行基因测序,利用NCBI数据库中提供的Nucleotide BLAST功能根据测序结果进行搜索,结果显示重组质粒pET28a-gdh上克隆的基因片段与枯草芽孢杆菌9902的葡萄糖脱氢酶基因序列(登录号为EF626962.1)之间的相似度和覆盖程度均达到100%。
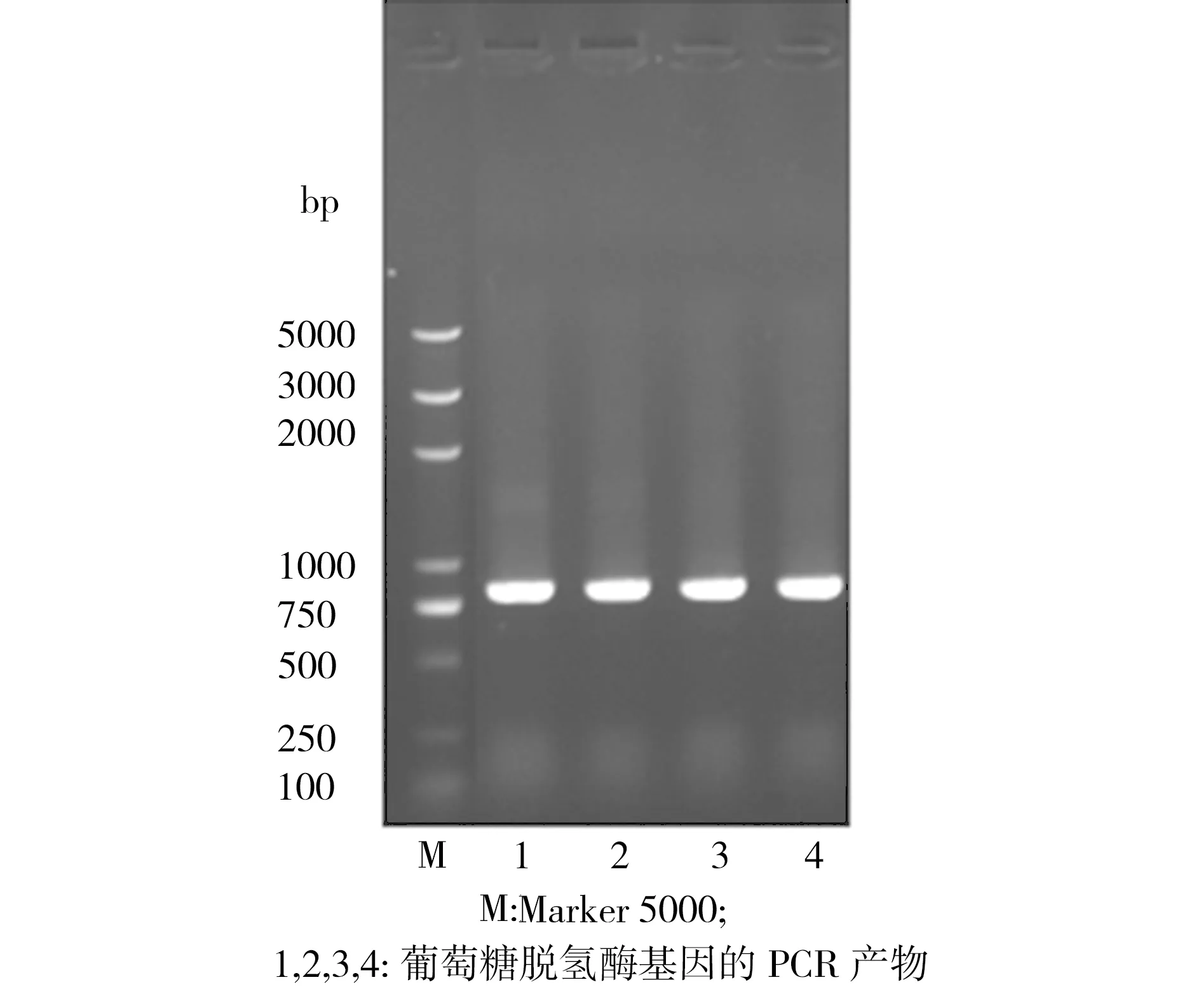
图1 PCR扩增产物电泳图
Fig.1 Electrophoretogram of PCR amplification products
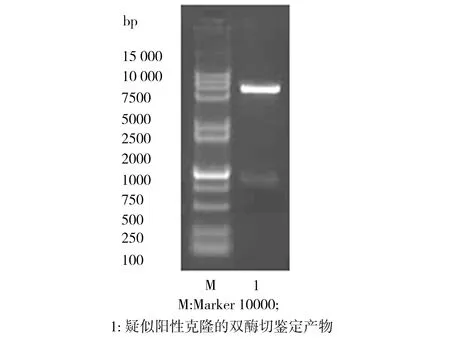
图2 疑似阳性克隆的双酶切鉴定产物电泳图
Fig.2 Electrophoretogram of the suspected positive clone by double enzyme digestion
2.2 重组葡萄糖脱氢酶的表达
利用SDS-PAGE检测经IPTG诱导后的重组菌(E.coliBL21(pET28a-gdh))中葡萄糖脱氢酶的表达情况,如图3所示。考虑到葡萄糖脱氢酶的分子大小约为28.8 kDa,加上表达蛋白中含有质粒pET28a的His-tag标签,预计表达的重组葡萄糖脱氢酶的相对分子量约为33 kDa。实验选择的阴性对照有:未经过和经过IPTG诱导的E.coliBL21(pET28a)以及未经IPTG诱导的E.coliBL21(pET28a-gdh)。由图3可知,同阴性对照相比,经IPTG诱导后的E.coliBL21(pET28a-gdh)检测结果中,在相对分子量35.0 kDa附近有明显的葡萄糖脱氢酶表达条带,此点与目标蛋白的相对分子量33 kDa一致,表明葡萄糖脱氢酶在重组菌中高效表达。根据BandScan 5.0软件分析SDS-PAGE图,可知葡萄糖脱氢酶(图3中的孔道4)的表达量占重组菌总可溶性蛋白质的64%。

图3 重组葡萄糖脱氢酶的SDS-PAGE检测结果
Fig.3 SDS-PAGE result of recombinant glucose dehydrogenase
将重组菌E.coliBL21(pET28a-gdh)的粗酶液在适当体系(pH7.0)中37 ℃反应30 min(以E.coliBL21(pET28a)作为阴性对照),测定反应体系在340 nm处的吸光值变化,获得酶催化反应的进程曲线。根据曲线中0~2 min内线性阶段的斜率可得出吸光值的变化率ΔA=0.75 min-1,结合NADPH标准曲线拟合的公式(y=0.004 83x+0.004 53,R2=0.999 82,其中y为340 nm处的吸光值,x为NADPH浓度(μmol/L)),可计算出单位时间内NADPH的浓度变化情况,从而得出粗酶液的酶活为23 291.95 U/L,而阴性对照的酶活仅为50.84 U/L。
采用Bradford法测定该重组菌E.coliBL21(pET28a-gdh)破胞上清液中的蛋白质浓度,根据BSA标准品的浓度与吸光值之间的标准曲线(y=0.005 79x-0.064,R2=0.997 01,y为595 nm处的吸光值,x为蛋白质含量(μg/mL)),测得其中蛋白质浓度为168.91 μg/mL,由此得出粗酶液的比酶活为137.90 U/mg,而阴性对照的蛋白质浓度为63.90 μg/mL,比酶活只有0.80 U/mg。
目前,很多研究人员测定了在大肠杆菌中异源表达枯草芽孢杆菌的葡萄糖脱氢酶的酶活特性。例如:周丽萍等[10]利用重组葡萄糖脱氢酶的初提液测定的比酶活为10 U/mg;徐军等[11]通过优化表达条件将重组酶的比酶活从10 U/mg提高到97 U/mg;Qiao等[12]构建的重组葡萄糖脱氢酶的比酶活为7.8 U/mg;徐娴等[13]通过优化诱导时间、温度等条件得出重组葡萄糖脱氢酶的比酶活为9.65 U/mg;李鸣等[14]构建的重组菌破碎细胞上清液葡萄糖脱氢酶比酶活为65.7 U/g。本课题组前期构建了克隆枯草芽孢杆菌AB90008中葡萄糖脱氢酶基因的重组大肠杆菌,其中酶的第124位残基上发生丝氨酸→半胱氨酸的突变,粗酶液的比酶活仅为18.4 U/mg[7]。为了获得更适合环己酮还原体系辅酶再生应用的酶活较高的重组菌,本研究重新选择枯草芽孢杆菌168作为葡萄糖脱氢酶基因的供体菌,从上述检测结果来看,重组菌表达的葡萄糖脱氢酶的酶活和比酶活要远远高出其他报道及本课题组前期构建的重组菌的相应数据。
2.3 重组葡萄糖脱氢酶的辅酶再生性能
E.coliBL21 (pET28a-eryKR1)中的酮还原酶域EryKR1以NADPH为氢供体还原环己酮,生成环己醇,在此过程中NADPH被氧化成NADP+。要想此反应能够循环反复,就必须往反应体系中不断添加还原型辅酶NADPH或是完成NADP+的再次还原(即还原性辅酶的再生)。重组菌E.coliBL21(pET28a-gdh)表达的葡萄糖脱氢酶能够通过氧化葡萄糖将NADP+还原成NADPH。
为了检测重组葡萄糖脱氢酶在环己酮还原反应体系中的辅酶再生性能,将E.coliBL21 (pET28a-eryKR1)和E.coliBL21(pET28a-gdh)双重组菌耦合进行环己酮的发酵,利用气相色谱检测发酵液中环己酮和环己醇的浓度(见图4),从而计算出相应的环己醇产率(见表1)。
由表1可知,当环己酮还原反应系统中只加E.coliBL21 (pET28a-eryKR1)和NADPH(第4组)时,环己醇产率很低,仅为5.22%;而当体系中加入E.coliBL21 (pET28a-eryKR1)、E.coliBL21(pET28a-gdh)、葡萄糖和NADPH(第1组)时,环己醇的产率高达82.21%。以E.coliBL21(pET28a)还原环己酮的反应体系作为阴性对照,其中几乎检测不出环己醇的含量。由于大肠杆菌中生成的NADPH较少,还原再生NADPH的能力很弱,因此,对于第3组和第4组没有添加GDH辅酶再生系统的环己酮还原体系,环己醇产率和环己酮转化率均很低,表明靠一次性添加外源性的NADPH无法从根本上满足环己酮还原反应对氢供体的需求。通过第1组和第3组的对比可知,E.coliBL21(pET28a-gdh)的加入可使环己醇的产率提升2.23倍;第2组的数据表明,即使不添加外源性NADPH,利用大肠杆菌宿主菌中生成的少量NADPH,GDH也能完成辅酶再生循环,为环己酮还原反应源源不断地提供氢供体;对比第1组和第2组的数据可知,外源性NADPH的加入可以使环己醇的产率提升22.5%左右,因此在存在GDH辅酶再生系统的条件下,额外地添加NADPH可以使得辅酶再生循环的基数提高,导致氢供体的增长速率加快,进而提高环己醇的产率。综上所述,本实验构建的重组菌E.coliBL21(pET28a-gdh)表达的葡萄糖脱氢酶可以解决E.coliBL21 (pET28a-eryKR1)催化环己酮还原体系的辅酶再生问题。
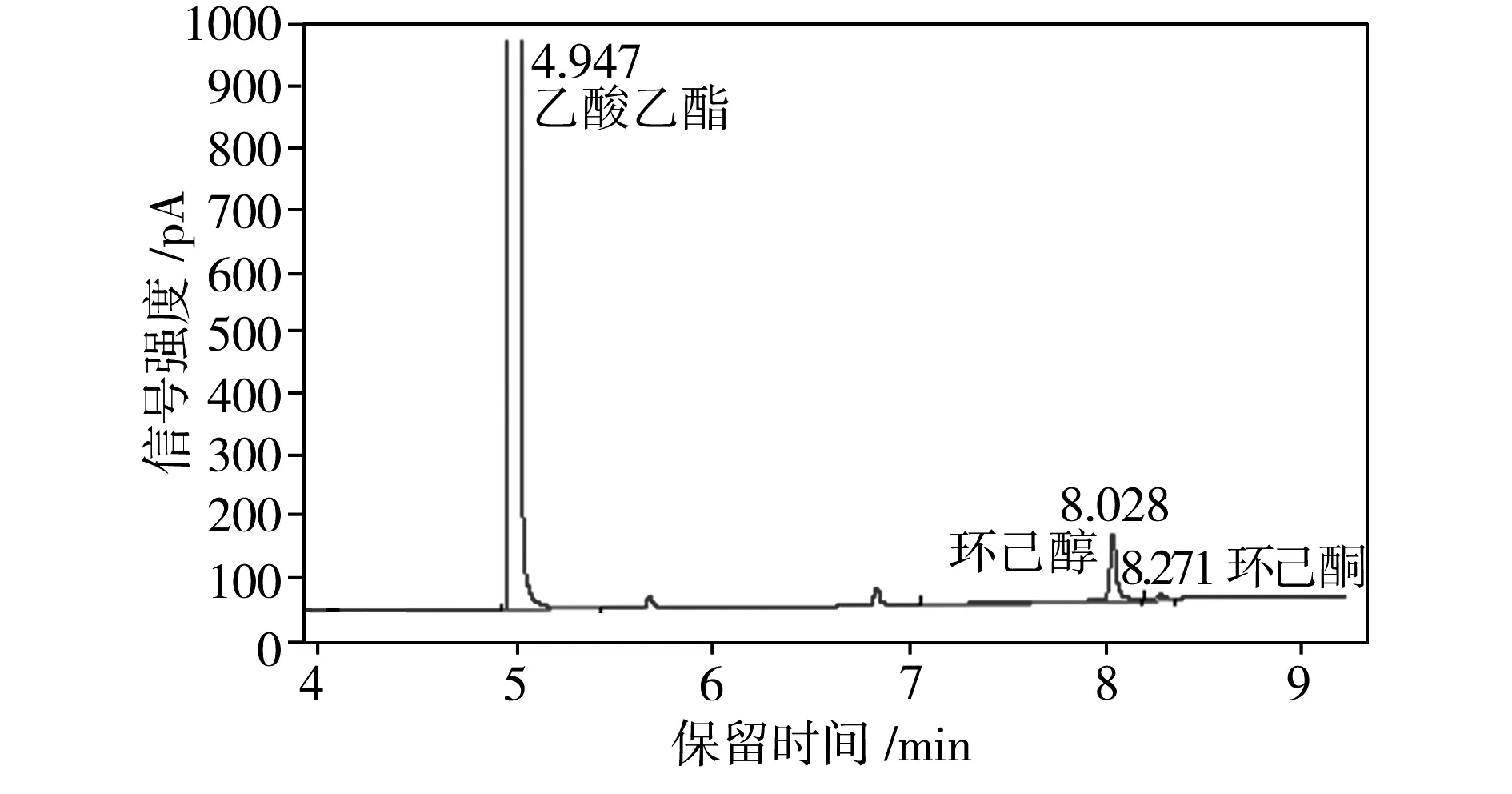
(a)第1组:E.coliBL21 (pET28a-eryKR1)+E.coliBL21(pET28a-gdh)+葡萄糖+NADPH
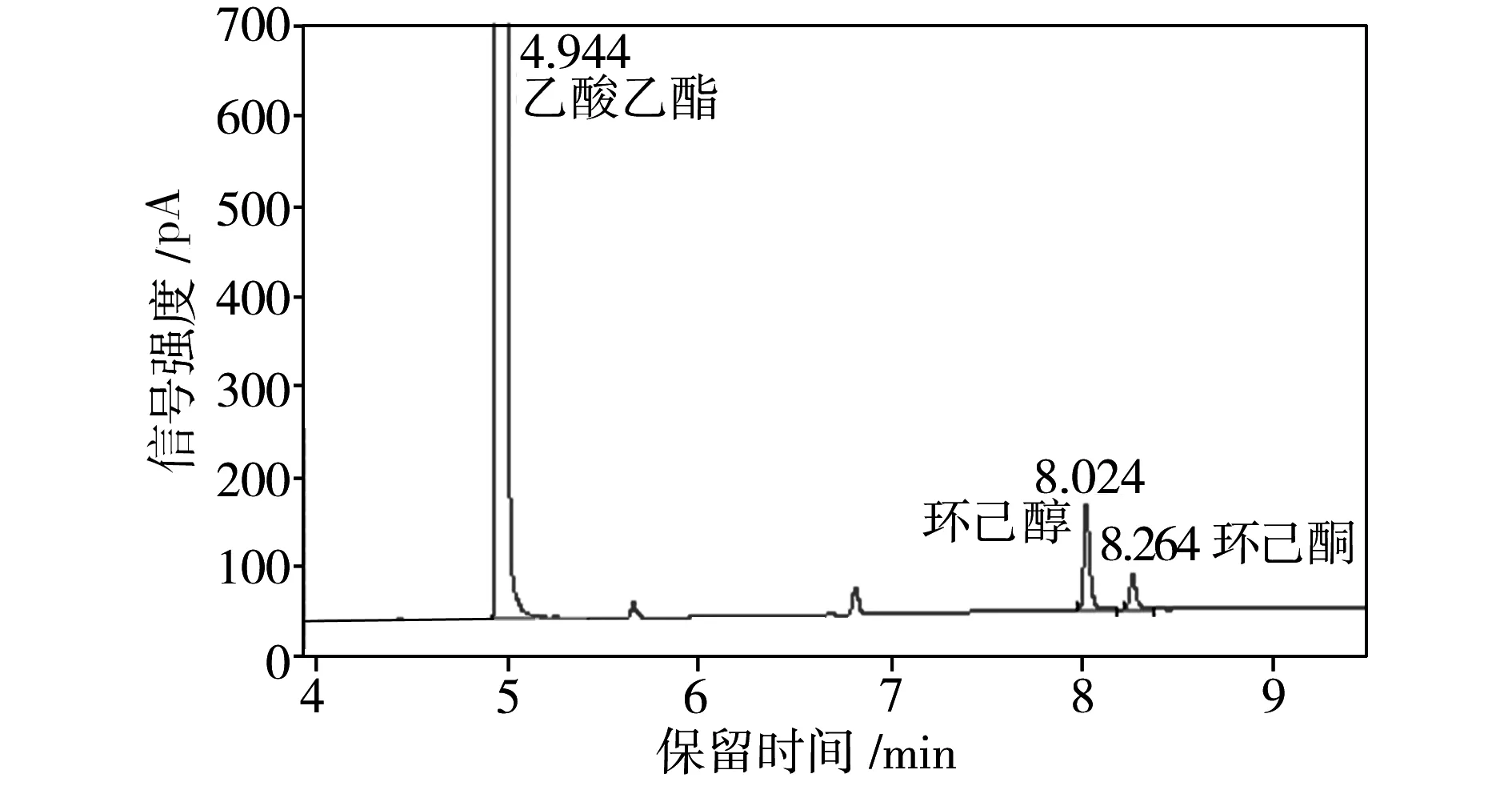
(b)第2组:E.coliBL21 (pET28a-eryKR1)+E.coliBL21(pET28a-gdh)+葡萄糖
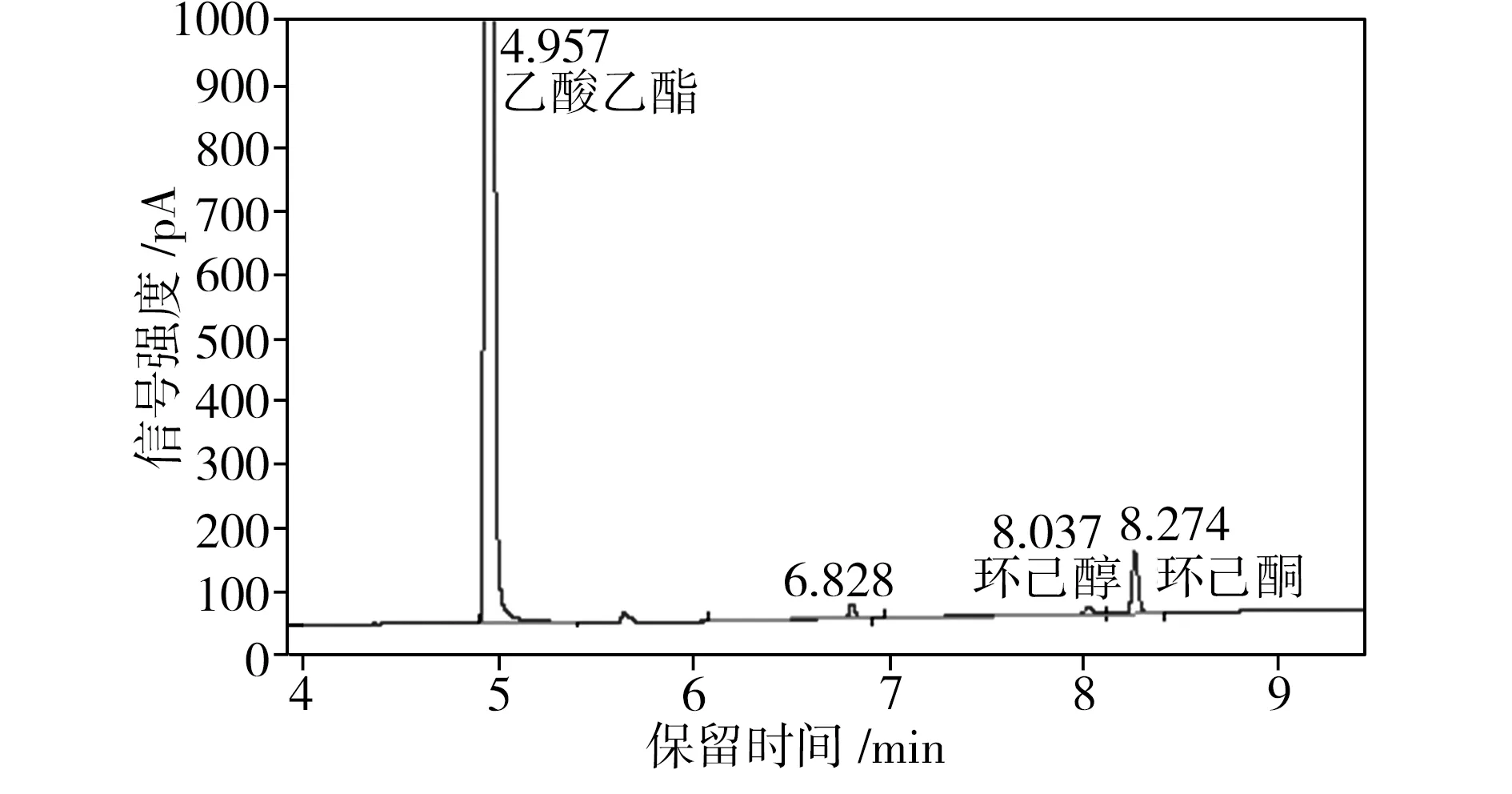
(c)第3组:E.coliBL21 (pET28a-eryKR1)+葡萄糖+NADPH
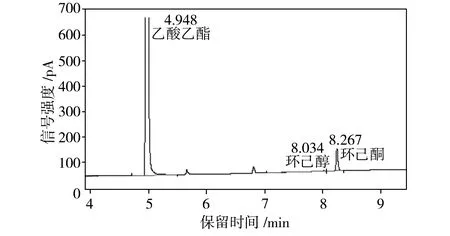
(d)第4组:E.coliBL21 (pET28a-eryKR1)+ NADPH
图4 环己酮发酵液的气相色谱图
Fig.4 Gas chromatography of cyclohexanone fermentation brothes
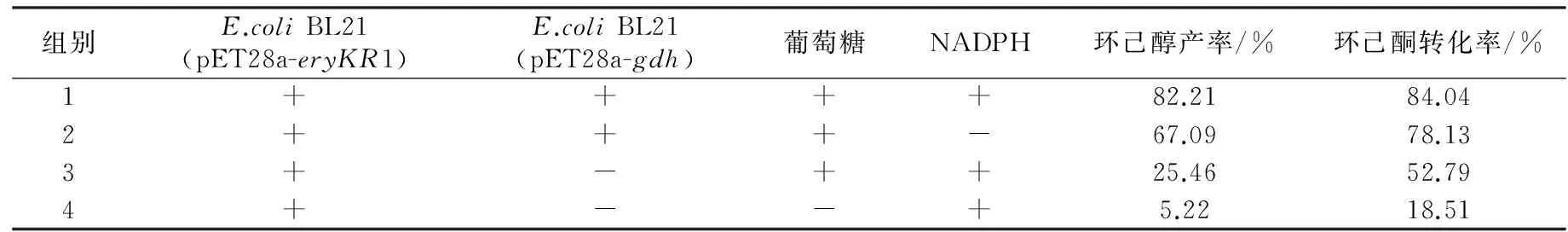
表1 重组葡萄糖脱氢酶在环己酮还原体系中的辅酶再生性能检测结果
利用基因工程技术实现葡萄糖脱氢酶的高效表达,并构建葡萄糖脱氢酶与羰基还原酶耦联的辅酶循环系统,可以解决生物催化潜手性羰基不对称还原中的辅酶再生问题。目前,这一辅酶再生方法已经被广泛地应用到很多手性化合物的合成中。例如:一些研究人员构建的葡萄糖脱氢酶与羰基还原酶耦联的辅酶再生体系可以催化4-氯乙酰乙酸乙酯还原生成4-氯-3-羟基丁酸乙酯,其中产率最高可以达到99%[6,15-16];Gao等[17]完成了枯草芽孢杆菌葡萄糖脱氢酶和氧化葡糖杆菌NADPH依赖的羰基还原酶在同一大肠杆菌中耦联表达,这种一菌双酶的辅酶再生系统可以在75 min内还原14.3 g/L双乙酰生成12.2 g/L (S)-3-羟基丁酮;Ema等[18]实现了酿酒酵母羰基还原酶和巨大芽孢杆菌葡萄糖脱氢酶的共表达,合成了20种醇,其中(S)-2-羟基-4-辛酮的产率为71%;李鸣等[14]共表达了羰基还原酶和葡萄糖脱氢酶,建立了双酶耦联反应,其中产物(R)-4-甲氧基-1-苯乙醇的产率高达82%;宿宇宁等[19]将来源于枯草芽孢杆菌的羰基还原酶和葡萄糖脱氢酶在大肠杆菌中共表达,对10 g/L的2-羰基-4-苯基丁酸乙酯进行不对称还原,转化率大于99%;欧阳健等[20]将羰基还原酶与葡萄糖脱氢酶耦联再生NADPH不对称合成(S)-1-苯基-1,2-乙二醇,转化率达到90.91%。本研究将构建的异源表达葡萄糖脱氢酶的重组大肠杆菌添加到克隆了酮还原酶域基因的重组菌的环己酮还原反应体系中,环己醇产率可以达到82.21%。根据本课题组前期的研究成果,此双重组菌耦联体系可以还原潜手性脂环酮(如2-甲基环己酮、1,2-环己二酮等)[21-22],说明葡萄糖脱氢酶和羰基还原酶的耦联除了可以应用到4-氯-3-羟基丁酸乙酯等重要手性砌块的合成中之外,还有望为工业生产药物中间体——含环己基的手性醇解决辅酶再生问题。
3 结论
(1)构建了异源表达枯草芽孢杆菌葡萄糖脱氢酶的重组大肠杆菌,其中克隆的葡萄糖脱氢酶基因gdh经Nucleotide BLAST功能分析显示与枯草芽孢杆菌9902的gdh基因(登录号为EF626962.1)的序列一致性达到100%。
(2)实现了重组型葡萄糖脱氢酶的高水平表达,表达量占全菌可溶性蛋白质的64%,且粗酶液的比酶活达到137.90 U/mg。
(3)建立了异源表达葡萄糖脱氢酶和酮还原酶域的双重组菌耦合还原环己酮的反应体系,其中环己醇的产率高达82.21%。由此可解决环己酮还原反应中的辅酶再生问题,促进酮还原酶域在潜手性脂环酮不对称还原中的工业应用。
[1] 彭益强,张曙伟.手性化合物酶法制备中辅酶再生体系的构建与应用进展[J].化工进展,2014,33(7):1826-1831.
[2] 王秋雨,钦传光,左小佳,等.伴有辅酶再生的生物催化过程[J].化学通报,2009,72(7):587-593.
[3] 周丽萍,徐军.巨大芽孢杆菌葡萄糖脱氢酶基因的重组和表达研究[J].中国现代医学杂志,2007,17(2):172-174.
[4] 王文溪,葛欣,韩月梅,等.甲基营养菌MP688葡萄糖脱氢酶基因分离鉴定及性质研究[J].生物技术通讯,2013,24(6):805-809.
[5] 戴宝新,冯惠勇,李天明,等.利用TAIL-PCR克隆Gluconobactersuboxydans葡萄糖脱氢酶基因及其生物信息学分析[J].食品科学,2014,35(1):194-198.
[6] 余涛,胡蝶,邬敏辰,等.重组葡萄糖脱氢酶的酶学性质及其偶联辅酶再生[J].食品与生物技术学报,2014,33(9):910-916.
[7] 李凌凌,吕早生,吴敏,等.重组的葡萄糖脱氢酶催化辅酶的再生性质[J].华中科技大学学报:自然科学版,2010,38(3):112-115.
[8] 杨忠华,左振宇.生物工程专业实验[M].北京:化学工业出版社,2014:65-73.
[9] 韩增叶,孙继国,葛喜珍,等.大肠杆菌葡萄糖脱氢酶基因的克隆与原核表达[J].北京化工大学学报:自然科学版,2012,39(1):68-71.
[10]周丽萍,赵燕,王卉放,等.枯草芽孢杆菌葡萄糖脱氢酶的克隆和表达[J].江苏大学学报:医学版,2004,14(1):7-10.
[11]徐军,周丽萍.重组葡萄糖脱氢酶基因表达条件的优化及其稳定性[J].江苏医药,2008,34(7):712-714.
[12]Qiao J J, Lu F P, Chen Q M, et al. Cloning and high level expression of glucose dehydrogenase gene fromBacillussubtilisinE.coli[J].南开大学学报:自然科学版, 2004, 37(2): 13-17.
[13]徐娴,谢承佳,何冰芳.枯草芽孢杆菌葡萄糖脱氢酶基因的克隆及高效表达[J].食品与生物技术学报,2007,26(5):75-78.
[14]李鸣,聂尧,张荣珍,等.氧化还原酶共表达耦联催化合成(R)-4-甲氧基-1-苯乙醇[J].食品与生物技术学报,2016,35(6):569-576.
[15]Ye Q, Cao H, Mi L, et al. Biosynthesis of (S)-4-chloro-3-hydroxybutanoate ethyl usingEscherichiacolico-expressing a novel NADH-dependent carbonyl reductase and a glucose dehydrogenase[J]. Bioresource Technology, 2010, 101(22): 8911-8914.
[16]Ni Y, Li C X, Wang L J, et al. Highly stereoselective reduction of prochiral ketones by a bacterial reductase coupled with cofactor regeneration[J]. Organic and Biomolecular Chemistry, 2011, 9(15): 5463-5468.
[17]Gao C, Zhang L, Xie Y, et al. Production of (3S)-acetoin from diacetyl by using stereoselective NADPH-dependent carbonyl reductase and glucose dehydrogenase[J]. Bioresource Technology, 2013, 137(6): 111-115.
[18]Ema T, Yagasaki H, Okita N, et al. Asymmetric reduction of ketones using recombinantE.colicells that produce a versatile carbonyl reductase with high enantioselectivity and broad substrate specificity[J]. Tetrahedron, 2006, 62(26): 6143-6149.
[19]宿宇宁,倪晔,王骏超,等.不对称还原制备光学纯(R)-2-羟基-4-苯基丁酸乙酯的双酶共表达重组菌的构建[J].催化学报,2012,33(10):1650-1660.
[20]欧阳健,徐岩,穆晓清,等.加强辅酶循环再生实现不对称合成(S)-1-苯基-1,2-乙二醇[J].四川大学学报:自然科学版,2007,44(4):861-866.
[21]李凌凌,吕早生,关海燕,等. 糖多孢红霉菌酮还原酶域在羰基还原中的应用[J].华中科技大学学报:自然科学版,2011,39(2):72-75.
[22]李尧益,李凌凌,吕早生,等.异源表达酮还原酶的重组菌单羰基还原1,2-环己二酮[J].广州化工,2016,44(18):62-65.
[责任编辑 尚 晶]
Expression of recombinant glucose dehydrogenase and its application to coenzyme regeneration
LiLingling,LiuYaoning,LvZaosheng,YangZhonghua,ZuoZhenyu,SongCaiwei
(College of Chemistry and Chemical Engineering, Wuhan University of Science and Technology, Wuhan 430081, China)
To solve the regeneration of hydrogen donor NADPH for cyclohexanone reduction catalyzed by the recombinant strainEscherichiacoliBL21 (pET28a-eryKR1) heterologously expressing ketoreductase domain (EryKR1), the recombinant strainE.coliBL21 (pET28a-gdh) was constructed which cloned the glucose dehydrogenase(GDH) genegdhfromBacillussubtilis. BLAST analysis showed that nucleotide sequence of the genegdhin the recombinant strain had the consistency of 100% with that ofB.subtilisstrain 9902 (accession number EF626962.1). SDS-PAGE analysis revealed that GDH could be highly expressed in the recombinant strain induced by IPTG, which accounted for 64% of the total soluble protein. Specific activity of GDH crude enzyme was 137.90 U/mg. Recombinat strainE.coliBL21(pET28a-gdh) was added to the cyclohexanone reduction reaction catalyzed byE.coliBL21(pET28a-eryKR1). Gas chromatography analysis showed that the yield of cyclohexanol in the two-recombinant strain reaction system was 82.21% and 3.23 times that of the system withoutE.coliBL21(pET28a-gdh), which confirmed that recombinant GDH could solve the coenzyme regeneration problem for cyclohexanone reduction catalyzed by ketoreductase domain.
glucose dehydrogenase; recombinant strain; heterologous expression; coenzyme regeneration; biocatalysis; cyclohexanone;Bacillussubtilis
10.3969/j.issn.1674-3644.2017.02.014
2016-12-26
国家自然科学基金资助项目(21376184);武汉科技大学青年科技骨干培育计划项目(2016XZ014);武汉科技大学大学生科技创新基金研究项目(16ZRA044).
李凌凌(1981-),女,武汉科技大学副教授,博士. E-mail:moonletsmile@163.com
Q789;TQ925
A
1674-3644(2017)02-0155-06
