脑脊髓病样表现的生物素酶缺乏症1例报告并文献复习
2017-04-12马秀伟辜蕊洁封志纯
马秀伟 侯 豫 辜蕊洁 封志纯
陆军总医院附属八一儿童医院神经发育科(北京 100700)
脑脊髓病样表现的生物素酶缺乏症1例报告并文献复习
马秀伟 侯 豫 辜蕊洁 封志纯
陆军总医院附属八一儿童医院神经发育科(北京 100700)
目的探讨脑脊髓病样表现的生物素酶(BTD)缺乏症的诊断和治疗。方法回顾分析1例BTD缺乏症患儿的临床资料,并分析相关文献。结果患儿,男,6岁,入院前3个月进行性双下肢痉挛性瘫痪;既往在3岁感冒后出现类似情况;平时手易脱皮,有口角炎。外院检查示视听诱发电位异常。入院后查脑脊液正常,头颅磁共振成像(MRI)示双侧枕叶、基底节区多发点片状稍长T1长T2异常信号;入院后出现延髓麻痹表现,给予气管插管呼吸机辅助通气。尿气相色谱-质谱(GC/MS)分析,尿乳酸、3-羟基异戊酸、3-甲基巴豆酰甘氨酸、甲基枸橼酸及3-羟基丙酸排泄量增高;血串联质谱技术(MS/MS)分析,丙酰基肉碱、3-羟基异戊酰肉碱(C5-OH)浓度明显升高;血BTD活性明显降低0.076 pmol/(min•mm3),确诊为BTD缺乏症。给予生物素40 mg/d口服,3天后撤机成功,2周后可行走,皮疹消退;3周后复查头颅MRI示原病灶消失,脊髓MRI未见异常。PCR直接测序法检测BTD基因,发现患儿第2号外显子上T172T/C杂合突变和第4号外显子上T1413C纯合突变,家系验证及数据库查询证实后者为致病性突变。出院后继续给予生物素口服20 mg/d,随访2年无异常。结论BTD缺乏症表现复杂多样,尿GC/MS及血MS/MS分析可协助诊断,BTD活性测定及BTD基因检测可进一步确诊此病,及时给予生物素治疗疗效显著。
生物素酶缺乏; 生物素; 脑脊髓病; 基因突变
多种羧化酶缺乏症(multiple carboxylase deficiency,MCD)是一种常染色体隐性遗传性先天代谢性疾病,由于支链氨基酸代谢过程中全羧化酶合成酶(holocarboxylase synthetase,HLCS,包括丙酰辅酶A羧化酶、3-甲基巴豆酰辅酶A羧化酶、丙酮酸羧化酶及乙酰辅酶A羧化酶)缺陷或其辅酶-生物素酶(biotinidase,BTD)缺陷所致,引起一系列神经、皮肤损害与代谢紊乱。BTD缺乏症在我国少于HLCS缺乏症[1],一般起病较晚,临床上以神经系统和皮肤损害为特征,但表现更为复杂多样,尤其在神经系统可出现瘫痪、惊厥、发育迟缓、视听障碍、意识障碍等,极易漏诊误诊,导致预后不良甚至死亡。若能早期确诊并给予生物素治疗,可迅速改善临床表现,往往预后良好。为提高临床医师对此病的认识,现将1例以脑脊髓病样表现的BTD缺乏症报道如下,并进行相关文献复习。
1 临床资料
患儿,男,6岁。因双下肢无力3个月入住八一儿童医院。患儿入院前3个月开始出现双下肢无力,走路不稳,容易摔跤,未予重视。入院前2个月逐渐不能独自行走,于当地医院就诊,查脊髓及头颅核磁共振成像(magnetic resonance imaging,MRI)、肌电图、生化全项、重金属检测、血尿便常规等均未见异常。视觉诱发电位示双侧P100潜伏期延长。听觉诱发电位显示左侧阈值60 dB,右侧阈值50 dB。给予营养神经及针灸等治疗,效果不佳,症状加重。病程中无发热、抽搐、意识障碍、头痛、呛咳、尿便障碍等,双上肢活动正常。既往在3岁感冒后出现类似双下肢无力情况,未明确原因,持续2个月后逐渐恢复正常。平时手容易脱皮,口唇及口角经常出现皮疹,局部用药后效果不佳。出生史无异常,精神运动发育正常。家族史无特殊,有一妹妹10个月,身体健康。
患儿入院体格检查:生命体征稳定,发育正常,口角暗红色皮疹、糜烂,双手指及手掌多处脱皮(图1)。心、肺、腹未见异常。神志清,精神可,颅神经检查未见异常,双上肢肌力肌张力正常,双下肢肌力3级,肌张力高,下肢腱反射亢进,双侧踝阵挛阳性,双侧巴氏征阳性,脑膜刺激征阴性。入院当日行脑脊液检查常规、生化均正常,血乳酸2.0 mmol/L,电解质、肝肾功能、肌酶、血氨均正常,头颅磁共振成像(MRI)检查显示:双侧枕叶、基底节区多发点片状稍长T1、长T2异常信号,液体衰减反转恢复序列FLAIR高信号,弥散加权成像DWI高信号(图2)。入院诊断:急性播散性脑脊髓炎,多发性硬化,遗传代谢病?入院当日下午患儿出现发热、咳嗽,晚间出现声音嘶哑、呛咳、呼吸费力、痰多、精神差、咽反射减弱。

图1 患儿体格检查
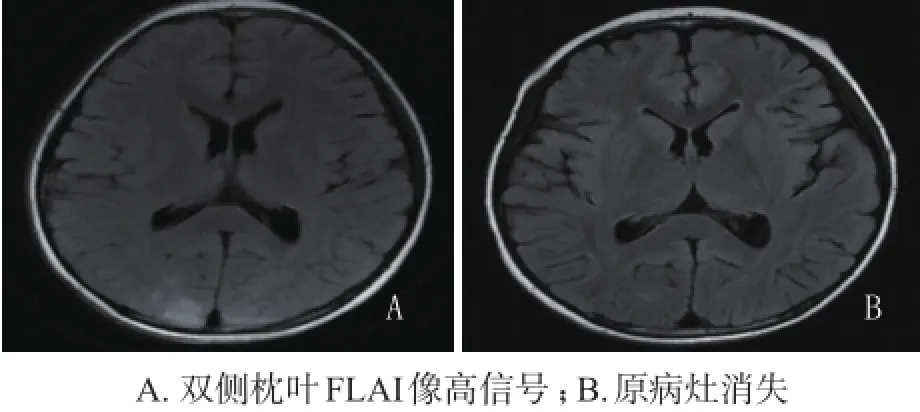
图2 患儿头颅MRI检查
入院后给予积极抗感染及静脉用丙种球蛋白滴注(intravenous immune globulin,IVIG)冲击治疗(总量2 g/kg),左卡尼汀、B族维生素等改善代谢。经药物治疗患儿症状无明显缓解,出现呼吸衰竭,二氧化碳分压(pCO2)76 mmHg,遂给予气管插管呼吸机辅助呼吸,并继续抗感染、改善代谢等治疗。
因患儿不除外遗传代谢病,入院后即进行血串联质谱分析MS/MS及尿气相色谱-质谱GC/MS检查。尿GC/MS检测示尿乳酸、3-羟基异戊酸、3-甲基巴豆酰甘氨酸、甲基枸橼酸及3-羟基丙酸排泄量增高。血MS/MS检测示丙酰基肉碱、3-羟基异戊酰肉碱(C5-OH)浓度明显升高。血浆BTD活性为0.076 pmol/ (min•mm3) [正常值6.3~9.3 pmol/(min•mm3)],确诊BTD缺乏症。给予生物素40 mg/d口服,患儿症状缓解,3天后撤机成功。继续口服生物素治疗,并进行康复训练,2周后患儿可自行行走,肌力恢复,病理征转为阴性,皮疹消失。3周后复查头颅MRI原病灶消失(图2),脊髓MRI未见异常,视觉诱发电位及听觉诱发电位均恢复正常。
经患儿家属签署知情同意书后,分别取患儿及其父母静脉血2 mL,提取DNA,利用Primer Premier 5.0设计引物,PCR产物进行测序分析,所得序列与UCSC数据库(http://genome.ucsc.edu/)中BTD基因序列进行对比。患儿BTD基因检测示第2号外显子上T172T/C杂合突变和第4号外显子上T1413C纯和突变,家系分析显示T172T/C杂合突变源于父亲,父母分别为4号外显子1413T>C杂合子(图3),考虑T1413C为致病性突变。出院后继续给予生物素口服20 mg/d。目前随访2年,患儿无不适,体格及精神运动发育均正常。
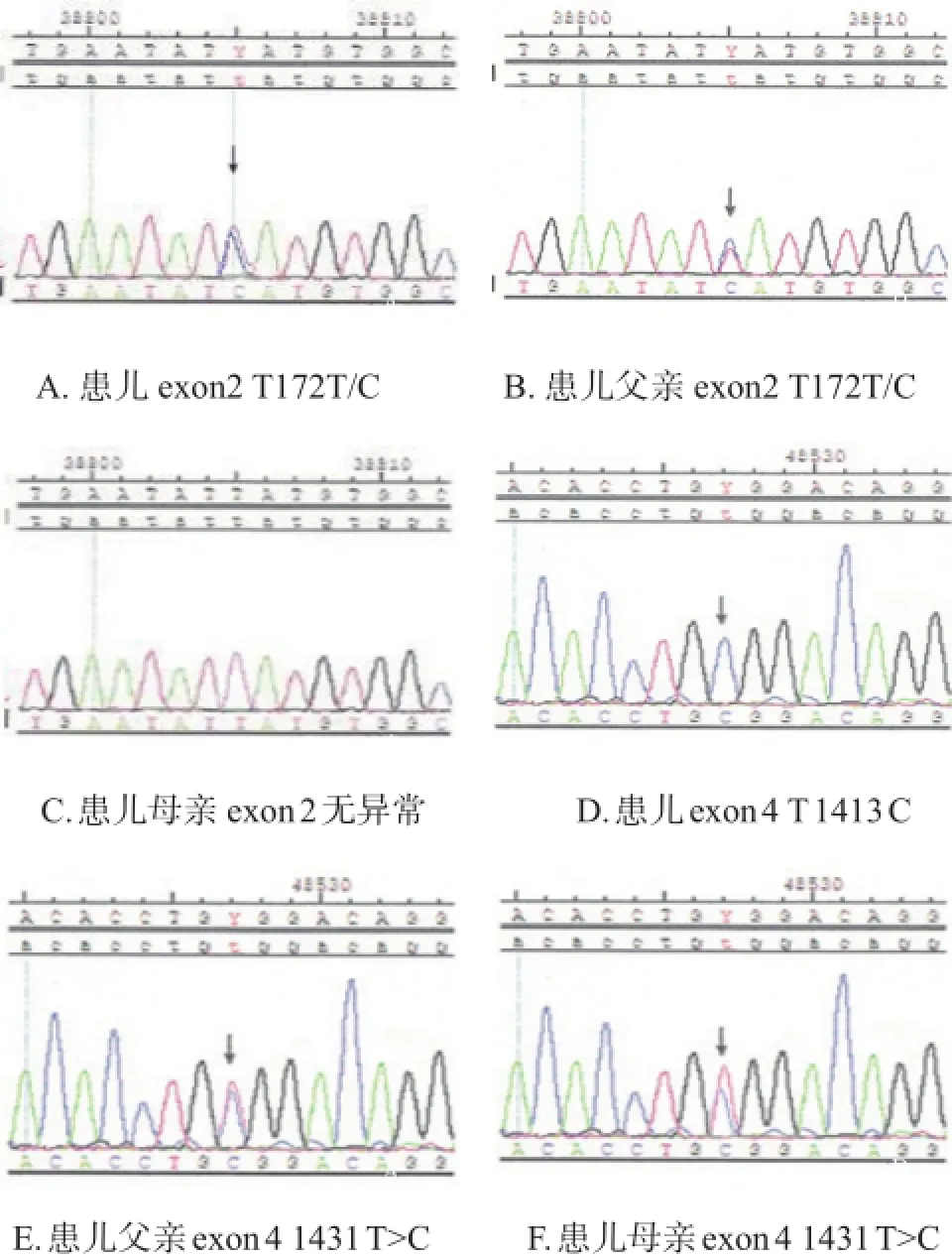
图3 基因测序图
2 讨论
生物素又称维生素B8、维生素H,是一种水溶性的含硫维生素,大部分从食物中摄取,少数在机体肠道中由细菌合成。生物素广泛存在于天然食物中,动物肝脏、蛋黄、鲜奶、大豆、酵母中含量高。食物中的生物素为蛋白结合状态,需在肠道经BTD作用生成游离的生物素才能发挥作用。正常机体中HLCS在ATP参与下催化生物素与羧化酶脱辅基蛋白结合,产生有活性的多种羧化酶,BTD还可将生物素从生物胞素和降解的羧化酶上裂解下来,使生物素再被利用。因此BTD缺乏可引起外源及内源性生物素均缺乏,4种相关羧化酶活性下降,乳酸、丙酮酸、3-羟基丙酸、丙酰甘氨酸、甲基枸橼酸、3-羟基异戊酸、甲基巴豆酰甘氨酸、巴豆酰甘氨酸等异常蓄积,能量合成障碍、肉碱消耗增加,引起一系列代谢紊乱与神经、皮肤损害。既往统计全球的BTD缺乏症发病率约1/60 000[2]。近年来许多国家已经对新生儿进行常规筛查,发现实际上的数字可能更高。匈牙利在1989—2008年期间对100多万名新生儿进行筛查,发现57例BTD缺乏症患儿,发病率为1/18 700[3],我国尚缺乏相关流行病学资料。
HLCS缺乏和BTD缺乏分别由HLCS基因和BTD基因突变所致,前者称为早发型(婴儿型)MCD,后者称为晚发型MCD。HLCS缺乏症多为新生儿至婴儿期起病,临床表现为喂养困难、呕吐、肌张力低下、惊厥、意识障碍、发育落后、皮疹、脱发等,急性期可合并代谢性酸中毒、高氨血症、低血糖等代谢紊乱,病死率高,相对容易识别。BTD缺乏症临床表现复杂,个体差异很大,可为神经系统、皮肤、呼吸系统、消化系统和免疫系统等多个系统非特异性损害。与HLCS缺乏症比较,BTD缺乏症常起病较晚,幼儿至成年各年龄段发病。本例患儿首次出现症状为3岁,且当时并未引起重视,可见早期识别此病相对困难。
BTD缺乏症的皮肤损害以脂溢性湿疹、过敏性皮炎、脱发、脱皮、结膜炎为多见,通常较轻微,但仍为BTD缺乏症重要表现之一。国内报道的18例BTD缺乏症中,16例有皮肤损害,皮肤湿疹样皮疹和/或皮肤干燥、粗糙[4]。本例患儿存在不明原因的口角炎、手脱皮等情况,给临床诊断此病提供了一定线索。BTD缺乏症神经系统损害更为明显且复杂多样,常表现为惊厥、肌张力障碍、痉挛性瘫痪、共济失调、发育迟缓、神经性耳聋、视神经萎缩等,可因发热、疲劳、饮食不当等诱发急性发作。本例患儿入院后出现发热、咳嗽,病情进行性加重,考虑与感染诱发病情加重有关。
部分BTD缺乏症患儿可有酸中毒和高乳酸血症,但代谢紊乱一般相对较轻。头颅和脊髓MRI可显示T1WI低信号、T2WI和FLAIR高信号,类似炎症或脱髓鞘脑脊髓病样的改变,易误诊为病毒性脑炎、多发性硬化、急性播散性脑脊髓炎、急性脊髓炎等,部分以白质损害为主的还可误诊为脑性瘫痪、脑白质营养不良等[5]。以脊髓病样表现的BTD缺乏症目前仍多为个案报道,以下肢瘫或四肢瘫为表现,伴或不伴皮疹及其他神经系统表现,脊髓MRI多出现异常信号,亦可显示正常,按脊髓炎给予IVIG及甲基泼尼松龙冲击治疗无效,但生物素治疗效果显著[6-9]。国外曾报道1例7岁的BTD缺乏症患儿出现四肢瘫痪,后又出现延髓麻痹表现,脊髓和脑干MRI均显示异常信号[8]。本例患儿以下肢瘫痪起病,伴有视听障碍,头颅MRI异常,且既往有类似发作,易误诊为脊髓道炎及多发性硬化。但患儿起病相对缓慢,持续时间长且病程中不伴随发热,脑脊液检查正常,给予免疫治疗无效,均不支持,而需要考虑是否存在遗传代谢性疾病,结合患儿特殊的皮疹,高度怀疑BTD缺乏症。本例患儿的特殊性更在于不单纯呈现脊髓病样表现,同时合并脑病样症状,出现精神差、视觉听觉障碍,且头颅MRI显示枕区和基底节区均有异常信号。BTD缺乏症的病理检查可有脑部半球深部灰质、脑干、脊髓的水肿和坏死,白质呈脱髓鞘[10],MRI类似炎症改变,但经过积极治疗可恢复,可予以鉴别诊断。本例患儿早期曾于外院进行头颅脊髓MRI检查均未见异常,病情进展时头颅MRI呈现异常信号,因病情进展迅速出现延髓麻痹,当时未能进行脊髓MRI检查,后期复查均正常,考虑可能当时存在水肿性病变,经过生物素治疗后恢复。
BTD缺乏导致糖、氨基酸、有机酸、脂肪酸等多种物质代谢障碍,线粒体能量合成障碍,肉碱消耗增加,典型患者尿GC/MS分析尿乳酸、丙酮酸、甲基枸橼酸、3-羟基丙酸、3-羟基异戊酸、3-羟基巴豆酰甘氨酸可明显升高。血MS/MS分析可显示丙酰基肉碱、3-羟基异戊酰肉碱浓度明显升高。目前血尿代谢病筛查已经在国内广泛普及,简便易行,可为此病诊断提供非常重要的线索,患者确诊需依赖血BTD活性测定。本例患儿干燥血滤纸片BTD活性测定<0.1 pmol/ (min·mm3),与文献报道一致[5]。BTD缺乏症临床上分为完全性缺乏和部分性缺乏,完全性缺乏是指血浆或血清BTD活性低于正常均值的10%,部分性缺乏是指酶活性介于正常均值的10%~30%[11]。部分性缺乏者症状可较轻,甚至终生不出现症状。本例患儿临床症状重,BTD活性低于正常值10%,属于完全性缺乏。
BTD基因检测亦可以确诊此病并有助于进行遗传咨询和再生育指导。BTD基因位于3p25,全长约为23 kb,由4个外显子和3个内含子组成。目前报道的BTD基因突变超过150余种,先证者多数为纯合突变或复合杂合突变[12,13]。在土耳其,常见的突变种类分别为位于第2外显子的c.235C>T和位于第4外显子的几种突变,分别为c.470G>A、c.557G>A、c.1330G>C、c.1368A>C、c.1489C>T、c.1595C>T[14]。有报道外显子2和4突变种类最多,1368 A>C引起 Q456H和双突变A171T:D444H为常见突变[3];也有报道最常见突变为c.1330G>C 引起的D444H[15]。此病为常染色体隐性遗传,父母亲多数为携带者,但亦有新生突变的报道[16]。本例患儿两处突变分别为外显子2的T172T/C杂合突变和外显子4的T1413C纯合突变,家系验证证实T172T/C来源于父亲,T1413C纯合突变分别来源于父亲母亲,父母均为杂合子。经文献检索,外显子4的T1413C已经有报道[17],考虑为致病性突变,对进一步研究BTD缺乏症基因型和表型的关系具有重要意义。
生物素治疗BTD缺乏症效果显著,起效迅速,推荐起始剂量10~40 mg/d,根据不同个体临床及实验结果调整用药量,即可终止疾病进展,修复已有的神经损害。患者需要终生补充生物素,左旋肉碱、B族维生素、中链脂肪酸等营养支持有助于纠正代谢紊乱及康复。本例患儿因病情危重,故起始剂量给予40 mg/d,病情稳定后改为20 mg/d,经随访一切正常,治疗效果好。国外报道患儿2个月内出现延髓麻痹和四肢痉挛性瘫痪,确诊BTD缺乏症后给予生物素20 mg/d,1周后延髓麻痹恢复,1个月后瘫痪症状明显改善,亦表明此类患儿如能及时确诊并给予生物素治疗,可迅速逆转病情,改善预后[8]。
目前有多个国家已经开展新生儿干血片BTD活性测定筛查BTD缺乏症,以便早期诊断并早期给予替代治疗,改善预后。对于患病者家庭可进行遗传咨询,再生育指导,避免此类患儿出生。
[1] 李秀珍, 刘丽, 盛慧英, 等. 多种羧化酶缺乏症15例临床分析及长期随访 [J]. 中华实用儿科临床杂志, 2014, 29(8):590-594.
[2] Wolf B. Disorder of biotin metabolism [M]// Scriver CR, Beaudet AL, Sly WS, et al. The metabolic and molecular basis of inherited Disease. 8th ed. New York, NY:McGraw-Hill, 2001: 3935-3962.
[3] Milankovics I, Nemeth K, Somogyi C, et al. High frequencies of biotinidase (BTD) gene mutations in the Hungarian population [J]. J Inherit Metab Dis, 2010, 3(Suppl 3): S289-S292.
[4] 王红梅,吴沪生.生物素酶缺乏症18例临床研究[J].中国实用儿科杂志, 2010, 25 (11):874-879.
[5] 包新华, 杨艳玲, 吴晔, 等. 多羧酶缺乏患儿的临床与生化特点研究[J]. 实用儿科临床杂志, 2003, 18(6): 423-425.
[6] Wiznitzer M, Bangert BA. Biotinidase deficiency: clinical and MRI findings consistent with myelopathy [J]. Pediatr Neurol, 2003, 29 (1): 56-58.
[7] Yang Y, Li C, Qi Z, et al. Spinal cord demyelination associated with biotinidase de fi ciency in 3 Chinese patients [J]. J Child Neurol, 2007, 22 (2): 156-160.
[8] Raha S, Udani V. Biotinidase deficiency presenting as recurrent myelopathy in a 7-year-old boy and a review of the literature [J]. Pediatr Neurol, 2011, 45 (4): 261-264.
[9] Komur M, Okuyaz C, Ezgu F, et al. A girl with spastic tetra paresis associated with biotinidase deficiency [J]. Eur J Paediatr Neurol, 2011, 15(6): 551-553.
[10] Honavar M, Janota I, Neville BG, et al. Neuropathology of biotinidase deficiency [J]. Acta Neuropathol, 1992, 84(4):461-464.
[11] Borsatto T, Sperb-Ludwig F, Pinto LL, et al. Biotinidase deficiency: clinical and genetic studies of 38 Brazilian patients [J]. BMC Med Genet, 2014, 15: 96.
[12] Wolf B. Biotinidase deficiency: “if you have to have an inherited metabolic disease, this is the one to have” [J]. Genet Med, 2012, 14 (6):565-575.
[13] Karaca M, Ozgul RK, Unal O, et al. Detection of biotinidase gene mutations in Turkish patients ascertained by newborn and family screening [J]. Eur J Pediatr, 2015,174 (8): 1077-1084.
[14] Kasapkara CS, Akar M, Ozbek MN, et al. Mutations inBTDgene causing biotinidase def i ciency: a regional report [J]. J Pediatr Endocrinol Metab, 2015, 28(3-4): 421-424.
[15] Couce ML, Perez-Cerda C, Garcia Silva MT, et al. Clinical and genetic findings in patients with biotinidase deficiency detected through newborn screening or selective screening for hearing loss or inherited metabolic disease [J]. Med Clin (Barc), 2011, 137(11): 500-503.
[16] Tonin R, Caciotti A, Funghini S, et al. Biotinidase def i ciency due to a de novo mutation or gonadal mosaicism in a first child [J]. Clin Chim Acta, 2015, 445: 70-72.
[17] Thodi G, Molou E, Georgiou V, et al. Mutational analysis for biotinidase deficiency of Greek patients’ cohort ascertained through expanded newborn screening [J]. J Hum Genet, 2011, 56(12):861-865.
(本文编辑: 邹 强)
Biotinase def i ciency manifested as encephalomyelopathy: a case report and literature review
ObjectiveTo explore the diagnosis and treatment of biotinase deficiency (BTD) manifested as encephalomyelopathy.MethodsThe clinical data of one child with BTD were retrospectively analyzed. The pertinent literatures were reviewed.ResultsA six-year-old male child suffered from progressive spastic paralysis of lower limbs for 3 months before admission. A similar symptoms occurred after a cold in 3-year-old. It was easy to peel skin on her hands and she had angular stomatitis. Audio visual evoked potential was detected to be abnormal in other hospital. After hospitalizion, the cerebrospinal fl uid examination was normal, and MRI showed long T1 long T2 signals bilateral occipital lobe and basal ganglia region . Because the child represented medulla palsy, and so the tracheal intubation ventilator was administrated to assist ventilation. Urine gas chromatography/mass spectrometry (GC/MS) analysis showed increases of lactic acid, 3-hydroxy acid, 3-tiglyl glycine, methylcitric acid, and ethylene lactic acid. Serum MS/MS analysis showed that the concentrations of propionyl carnitine and 3-hydroxyisovaleryl carnitine were increase obviously. The serum biotinase level was signif i cantly decrease to 0.076 pmol/( min•mm3). The diagnosis of BTD was conf i rmed. After supplementation biotin , 40 mg/d, the ventilator was successfully weaned on the third day, the child walked again after 2 weeks, and the rash was vanished. After 3 weeks, the head MRI showed disappearance of the original lesion, and there was no abnormal in spinal cord. TheBTDgene detected by PCR direct sequencing showed a heterozygosis mutation of T172T/C in the second exon and a homozygous mutation of T1413C in the fourth exon, which was conf i rmed as a pathogenic mutation by pedigree verif i cation and database query. After discharge, the oral administration of biotin 20 mg/d continued, and no abnormality was found in 2 years of follow-up.ConclusionsThe manifestations of BTD are complex and diverse. The analysis of urine GC/MS and serum MS/MS can assist the diagnosis. The determination of biotinase activity and gene detection ofBTDcan further conf i rm the diagnosis. Timely biotin supplementation has signif i cant treatment eff i cacy.
biotinase def i ciency; biotin; encephalomyelopathy; gene mutation
10.3969/j.issn.1000-3606.2017.01.010
2016-07-06)
MA Xiuwei, HOU Yu, GU Ruijie, FENG Zhichun (Department of Neurology and Development, Aff i liated Bayi Children’s Hospital, PLA Army General Hospital, Beijing 100700, China)
