婴儿期结节性硬化症合并郎格汉斯细胞组织细胞增多症并继发噬血细胞性淋巴组织细胞增生症1例报告
2017-04-12罗新辉
陈 芬 孙 岩 罗新辉
新疆维吾尔自治区人民医院(新疆乌鲁木齐 830000)
婴儿期结节性硬化症合并郎格汉斯细胞组织细胞增多症并继发噬血细胞性淋巴组织细胞增生症1例报告
陈 芬 孙 岩 罗新辉
新疆维吾尔自治区人民医院(新疆乌鲁木齐 830000)
目的探讨结节性硬化症(TSC)合并郎格汉斯细胞组织细胞增多症(LCH),并继发噬血细胞性淋巴组织细胞增生症(HLH)的诊断。方法回顾分析1例婴儿期诊断为TSC和LCH,且继发HLH患儿的临床资料。结果男性,1岁4个月,维吾尔族,生后4个月起病,以婴儿痉挛为首发症状,1岁时基因检测证实TSC2基因3-10号外显子杂合缺失,确诊为TSC的同时,患儿出现全身皮疹、发热、肝脾肿大和骨质缺损,经皮肤活检确诊为LCH,且又并发HLH。结论TSC合并LCH并继发HLH罕见,临床表现复杂,需要鉴别。
结节性硬化症; 郎格汉斯细胞组织细胞增多症; 噬血细胞性淋巴组织细胞增生症
结节性硬化症(tuberous sclerosis complex,TSC)是一种极为罕见的多系统受累的神经皮肤综合征,常染色体显性遗传[1]。郎格汉斯细胞组织细胞增生症(Langerhans cell histiocytosis,LCH)以单核巨噬细胞系统特定树突细胞和网状细胞增生为共同特点,可累及全身任意1个或多个器官,临床表现多样[2]。噬血细胞性淋巴组织细胞增生症(hemophagocytic lymphohistiocytosis,HLH)又称噬血细胞综合征,是一组由活化淋巴细胞和组织细胞过度增生引起的多器官高炎症反应临床综合征[3],分为原发性和继发性两大类,病情凶险,病死率极高。现报告1例共同患有TSC和LCH,且在病程中出现HLH患儿的临床特征及诊治经过,以提高临床医师对此类罕见疾病的认识。
1 临床资料
患儿,男性,维吾尔族,1岁4个月。生后即出现颜面部及躯干色素脱失斑并逐渐增大,4月龄时出现间歇性点头、双上肢抱球样上举、双眼上凝视和下肢屈曲,常成串发作,每次发作持续1~2 s,每组发作约7~10次,多于睡醒或入睡前发作。未曾就医。1岁时,患儿因频繁抽搐,进行头颅CT检查示双侧侧脑室旁多发致密结节影(图1),脑电图检查提示广泛慢波、棘慢波和多棘慢波发放,基因检测发现患儿存在TSC2基因3-10号外显子杂合缺失变异(图2),确诊为TSC。患儿父母及姐姐均未发现TSC2基因异常。采用丙戊酸钠、左乙拉西坦和氨已烯酸等对症治疗后,抽搐发作减少。在诊治TSC过程中,患儿出现间歇性发热、全身红色丘疹(可结痂,4月龄时也有类似皮疹,可自愈)和右侧外耳道流脓。X线提示颅骨及四肢长骨骨质破坏。皮疹活检病理及免疫组化报告(图3):可见郎格汉斯细胞,此类细胞CD1a+、Langerin+、S-100+、CD68+、Ki-67 30%+、CD117+,确诊为LCH。半月之后,患儿肝脾进行性增大。全血细胞计数减少,白细胞计数1.55×109/L,血红蛋白65 g/L,血小板计数33×109/L;三酰甘油2.13 mmol/L,血清铁蛋白1 017.5 ng/mL,可溶性CD25为7 927.2 pg/ mL;骨髓细胞学检查见到吞噬现象,临床诊断为HLH(与LCH有关)。按照国际组织细胞协会LCH Ⅲ方案治疗(包括泼尼松、长春花碱和依托泊苷),皮疹渐消退,血象恢复,目前仍在治疗中。

图1 患儿头颅CT

图2 患儿基因检测
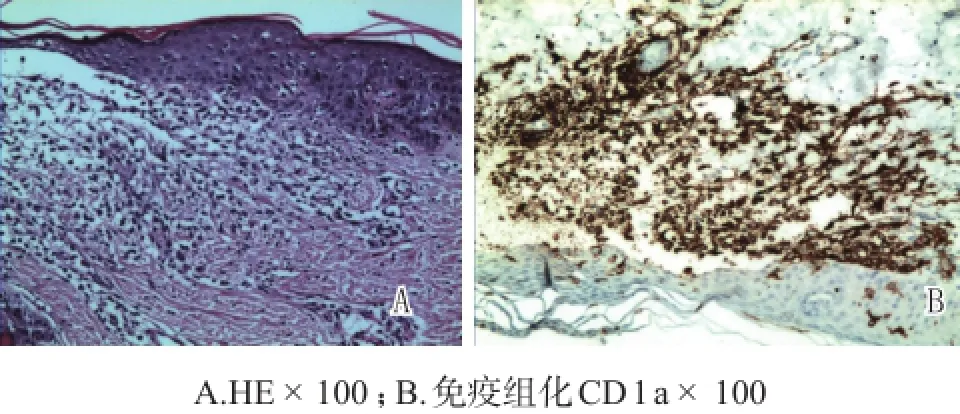
图3 患儿皮肤活检病理
2 讨论
TSC是一种常染色体显性遗传性疾病,发病率1/(10万~20万),全球约1 000 000例,中国约142 500~200 000例[4],仅1/3患儿有家族史[5]。本病常累及皮肤、脑、心、肾、肺、肝和眼等重要器官,是少数可以依靠临床表现就能诊断的遗传性疾病之一。随着研究的深入,2012年国际结节性硬化症共识会议对TSC的诊断标准进行了细化,并且确立基因诊断标准,即TSC致病基因分为TSC1(位于9q34)和TSC2(位于16p13.3)两种类型[6]。TSC2基因突变患者起病年龄更早且病情更为严重[7]。本例患儿存在TSC2基因突变,起病早,婴儿痉挛发作、室管膜下结节和多处皮肤色素脱失斑等临床特征均较为典型。
LCH是一组来源于骨髓朗格汉斯细胞的克隆性增生及聚集,伴有数量不等的中性粒细胞、嗜酸性粒细胞、淋巴细胞、浆细胞及多核巨细胞浸润,从而引起组织破坏的疾患[8]。国外报道年发病率为(0.35~0.70)/万,男女比例1.6~2.0∶1。该病临床表现差异极大,轻者仅累及皮肤或骨质,重者可累及多器官并造成重要脏器功能损害[9]。在存在LCH临床表现基础上,对病变组织活检进行组织学和免疫组化检查是确诊手段[10]。CD1a和Langerin阳性是确诊LCH的金标准。
TSC因TSC基因产物错构瘤蛋白或马铃薯球蛋白发生基因突变激活哺乳动物雷帕霉素靶蛋白(mTOR),导致患者脑组织、肾脏、心脏、肺和其他器官易发生错构瘤。意大利240例TSC患者中发现15例有错构瘤,其中7例为肾脏肿瘤,8例为非肾脏肿瘤[11]。研究还显示,TSC2基因突变组较TSC1基因突变组具有更高的肾血管平滑肌脂肪瘤和肾囊肿发生率[12,13]。TSC2基因的失活尤其是杂合TSC2突变,可能与肿瘤的发生发展有关[14]。检索中国知网及PubMed数据库未见同患TSC和LCH病例报道。本例患儿同时诊断TSC与LCH两种疾病,但两者从发生时间、发病机制及病理学上并无联系,因此考虑为两种疾病独立存在并无关联。TSC与LCH虽均可出现多器官受累,临床表现上有相似之处,但又不尽相同。皮疹,TSC患者可有面部血管纤维瘤、皮肤色素脱失斑和鲨革样斑;LCH患儿多为湿疹样、脂溢性皮炎样或紫癜样皮疹,皮损可出现于全身任何部位,以躯干、头面部多见。神经系统损害,TSC多以癫痫发作为首发症状并因此就诊,文献报道癫痫发生率为90%~96%[15],起病通常在1岁以内,可伴有神经系统发育和认知障碍[16,17],TSC合并癫痫的发作类型多样,1岁以内起病的患儿中约有1/3呈痉挛发作,并可伴有其他发作类型或与其他发作类型相互转变[18,19];LCH合并中枢神经系统病变的发病率为5%~24.5%[20],其机制可能与免疫反应基因尤其是人类主要组织相容性复合体有关[21],临床表现可有中枢性尿崩症、抽搐、生长发育迟缓、共济失调或智力倒退等[22]。骨质破坏,TSC可出现骨囊肿;LCH多有骨质破坏。
HLH是一类T细胞或 NK 细胞功能异常的免疫缺陷性疾病,分为原发性和继发性两大类。原发性HLH往往有家族遗传史或基因缺陷,异基因造血干细胞移植是唯一根治手段;继发性HLH多由感染、肿瘤、风湿免疫性疾病等引起,是基础疾病发展到一定程度所引起的一组临床综合征,预示基础疾病进入严重状态,治疗关键为控制基础疾病。国外报道,LCH合并HLH患儿的发病年龄均<1岁,且均为多器官受累的高危患儿,针对LCH治疗后HLH也可以得到控制[23,24]。但是LCH并发HLH者,总体预后较差。研究报道,12例合并HLH的LCH病例中,4例治疗无效死亡[25]。有学者认为,LCH与HLH之间存在相互联系比相互独立的可能性大,倾向于HLH可以继发于LCH,但其发病机制不明确,可能与T淋巴细胞及细胞因子的紊乱相关。本例患儿LCH后出现HLH,按LCH治疗后HLH很快好转,暂考虑为LCH继发HLH。但是,引起继发性HLH的疾病也可以促使原发性HLH发病,本例患儿虽无阳性家族史,但起病年龄小,在治疗过程中,若HLH出现复发,也应考虑进行原发性HLH相关基因分子标志检测。
结节性硬化及LCH均可在1岁以内起病,且均可有抽搐、皮疹及骨质破坏,虽然共同患有两种罕见疾病的概率极低,但是对于复杂病例,临床医师仍需提高警惕,因为早期诊断和正确治疗是改善预后的根本措施。
[1] Ess KC. Tuberous sclerosis complex: everything old is new again [J]. J Neurodev Disord, 2009, 1(2):141-149.
[2] 朱佳,方建培. 郎格汉斯细胞组织细胞增生症的诊治进展 [J]. 国外医学儿科学分册, 2005, 11( 32): 383-385.
[3] Risma K, Jordan MB. Hemophagocytic lymphohistiocytosis:updates and evolving concepts [J]. Curr Opin Pediatr, 2012, 24(1): 9-15.
[4] 廖建湘. 结节性硬化症规范化诊治标准简介 [J]. 中国医学论坛报, 2013, 11: A17.
[5] Yates JR. Tuberous sclerosis [J]. Eur J Hum Genet, 2006, 14(10):1065-1073.
[6] Bissler J, Henske E. Tuberous sclerosis complex:genes, clinical featurous and therapeutics [M]. USA: Wiley-Blackwell, 2010.
[7] Dabora SL, Jozwiak S, Franz DN, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs [J]. Am J Hum Genet, 2001, 68(1): 64-80.
[8] Ohnishi K, Komohara Y, Sakashita N, et al. Macrophages in Langerhans cell histiocytosis are differentiated toward M2 phenotype: their possible involvement in pathological processes [J]. Pathol Int, 2010, 60(1): 27-34.
[9] Haupt R, Minkov M, Astigarraga I, et al.Langerhans cell histiocytosis (LCH) : guidelines for diagnosis, clinical workup, and treatment for patients till the age of 18 years [J]. Pediatr Blood Cancer, 2013, 60(2): 175-184..
[10] Jaffe R. The diagnostic histopathology of Langerhans cell histiocytosis [M]// Weitzmans E. Histiocytic disorders of children and adults. Cambridge University Press, 2005.
[11] Peron A, Vignoli A, La Briola F, et al. Do patients with tuberous sclerosis complex have an increased risk for malignancies? [J]. Am J Med Genet A, 2016, 170 (6): 1538-1544.
[12] Sancak O, Nellist M, Goedbloed M, et al. Mutational analysis of theTSC1andTSC2genes in a diagnostic setting: genotypephenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex [J]. Eur J Hum Genet, 2005, 13(6):731-741.
[13] Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis [J]. Lancet, 2008, 372(9639): 657-668.
[14] Aizawa Y, Shirai T, Kobayashi T, et al. The tuberous sclerosis complex model Eker (TSC2+/-) rat exhibits hyperglycemia and hyperketonemia due to decreased glycolysis in the liver [J]. Arch Biochem Biophys, 2016, 590:48-55.
[15] 文家伦, 廖建湘, 陈黎, 等. 小儿结节性硬化症合并癫痫的随访研究 [J]. 中国当代儿科杂志, 2009, 11(12):996-998.
[16] Curatolo P, Jòżwiak S, Nabbout R, et al. Management of epilepsy associated with tuberous sclerosis complex (TSC):clinical recommendations [J]. Eur J Paediatr Neurol, 2012, 16(6): 582-586.
[17] 李亚勤, 操基清, 杨娟, 等. 结节性硬化症家系临床特点与基因分析 [J]. 中国现代神经疾病杂志, 2012, 12(3):300-306.
[18] Holmes GL, Stafstrom CE,Tuberous Sclerosis Study Group. Tuberous sclerosis complex and epilepsy: recent developments and future challenges [J]. Epilepsia, 2007, 48(4): 617-630.
[19] Hsieh DT, Jennesson MM, Thiele EA. Epileptic spasms in tuberous sclerosis complex [J]. Epilepsy Res, 2013, 106(1-2):200-210.
[20] Cochrane LA, Prince M, Clarke K. Langerhans’ cell histiocytosis in the paediatric population:presentation and treatment of head and neck manifestations [J]. J Otolaryngol, 2003, 32(1): 33-37.
[21] Krishna H, Behari S, Pal L, et al. Solitary Langerhans-cell histiocytosis of the clivus and sphenoid sinus with parasellar and petrous extensions:case report and a review of literature [J]. Surg Neurol, 2004, 62(5): 447-454.
[22] Vaisebuh SR, Bryceson YT, Allen CE, et al. Updates on histiocytic disorders [J]. Pediatr Blood Cancer, 2014, 61(7):1329-1335.
[23] Póvoas MI, Luís PP, Esteves I, et al. Severe Langerhans cell histiocytosis in an infant: haemophagocytic syndrome association [J]. BMJ Case Rep, 2014, 21:2014.
[24] Dokmanovic L, Krstovski N, Jankovic S, et al. Hemophagocytic lymphohistiocytosis arising in a child with Langerhans cell histiocytosis [J]. Turk J Pediatr, 2014, 56(4):452-457.
[25] Favara BE, Jaffe R, Egeler RM, et al. Macrophage activation and hemophagocytic syndrome in Langerhans cell histiocytosis: report of 30 cases [J]. Pediatr Dev Pathol, 2002, 5(2): 130-140.
Tuberous sclerosis and Langerhans cell histiocytosis combined with secondary hemophagocytic lymphohistocytosis in infancy: a case report
CHEN Fen, SUN Yan, LUO Xinhui (Department of Padiatric Hematology, People's Hospital of Xinjiang Uyghur Autonomous Region, Urumqi 830000, Xinjiang, China)
ObjectiveTo explore the diagnosis of tuberous sclerosis (TSC) combined with Langerhans cell histiocytosis (LCH) and secondary hemophagocytic lymphohistocytosis (HLH).MethodsOne case diagnosed of TSC combined wiht LCH and secondary with HLH in infancy was retrospectively analyzed.ResultsOne year and 4-month-old Uyghur boy when he was 4-month-old, there was onset of infantile spasm. Gene detection was performed when he was one year old and showed the absence of the exon 3 to 10 of TSC2 gene. The diagnosis of TSC was conf i rmed. Meanwhile, the boy also suffered with skin rash all around, fever, hepatosplenomegaly, and bone defect. The diagnosis of LCH was conf i rmed by skin biopsy. In addition, the boy was complicated HLH.ConclusionsIt is rarely seen that TSC combined with LCH, and secondary HLH in one case. The clinical features were complex and need to be differetiate.
tuberous sclerosis; Langerhans cell histiocytosis; hemophagocytic lymphohistocytosis
10.3969/j.issn.1000-3606.2017.01.012
2016-07-17)
(本文编辑:蔡虹蔚)
罗新辉 电子信箱: xinhuil@ sohu.com.
