遗传性血管性水肿一家系六例报告
2017-04-06李春香
李春香
遗传性血管性水肿一家系六例报告
李春香
血管性水肿,遗传性;腹痛;误诊;咽炎
遗传性血管性水肿(hereditary angioedema, HAE)是以C1酯酶抑制因子(C1INH)或FⅫ基因突变为分子遗传学基础、以反复发作的皮肤和(或)黏膜下等软组织水肿为主要表现的遗传性疾病,临床较为罕见,易误诊。我院近期收治HAE 1例,对其进行家系调查发现家族中有6人罹患此病,确诊前多被误诊,现报告如下。
1 临床资料
1.1 病例1 男,52岁,先证者(家系中Ⅱ6)。因反复局限性皮肤水肿15年余,喉头水肿5年,水肿再次发作1 d入院。15年前无明显诱因出现反复双上肢远端大片皮肤水肿,伴红肿,无压痛,持续2~3 d可自行消退,以春冬季发作为主,病程初期每年发作1~2次,未引起重视,后发作频繁。5年前出现喉头水肿,因喉镜下喉头、会厌肿胀明显,咽部憋闷,予糖皮质激素冲击治疗,同时予抗过敏治疗,症状无改善,考虑急性咽炎、会厌炎伴急性喉头水肿,予气管切开后症状好转。此后喉头水肿反复发作,予对症治疗后均好转出院。1 d前再次出现左上肢远端皮肤水肿,为进一步诊治到我院就诊,以水肿原因待查收入院。自患病以来,无口腔溃疡反复发作,无多关节肿痛,无腰背痛,无口干、眼干,无皮疹,食欲可,尿便未见异常。否认肝炎、结核病等传染病病史,否认自身免疫性疾病。
查体:生命体征正常,心肺腹及神经系统检查未见阳性体征,左上肢远端局限性非凹陷性水肿。查血尿常规、肝肾功能、血糖、血脂、红细胞沉降率、抗链球菌溶血素“O”、类风湿因子及C反应蛋白均正常;抗双链DNA抗体、抗核抗体、抗U1RNP/Sm抗体、抗Sm抗体、抗SSA抗体、抗SSB抗体、抗Scl-70抗体、抗JO-1抗体及抗核糖体P蛋白均阴性;全套吸入性及食入性过敏原均阴性;补体C4为 0.065 g/L(正常参考值0.17~0.48 g/L)。考虑血管性水肿,建议转北京进一步诊治。后随访,在北京某医院查补体C1抑制因子0.04 g/L(正常参考值0.21~0.39 g/L),确诊为HAE,予达那唑0.2 g、2/d口服。2个月后复查补体C4水平恢复正常,后达那唑减为0.2 g/d维持治疗,未再出现喉头水肿,偶有四肢皮肤水肿时,予达那唑适当加量,临床症状明显改善。
1.2 病例2 女,61岁,家系中Ⅱ4。因反复腹痛30余年,再次出现腹痛入院。30余年前无明显诱因出现反复腹痛,无固定痛点,每年发作2~3次,多次以急腹症收入院,考虑肠梗阻、原发性腹膜炎,予对症处理2~3 d后缓解。4年前患脑梗死,遗有左侧肢体活动障碍。10个月前因反复胸痛、胸闷收入院,住院期间胸痛症状突然加重,心电图示:V1~6导联ST段抬高,Q波形成,考虑急性心肌梗死。行急诊冠状动脉(冠脉)造影示:左冠脉主干正常,左前降支高位D1段狭窄最重处为70%,近段完全闭塞,左回旋支未见明显狭窄及阻塞性病变,右冠脉第一转折处狭窄30%,中段见多处斑块,术中于左前降支植入支架1枚,术后再次出现胸痛,前壁导联未见明显回落,继之出现言语不清、嗜睡,转重症监护病房,经生命支持、抗凝、强心、利尿、活血化瘀等处理,症状好转出院。出院后第4天再次出现腹痛,入住我院普外科,考虑肠梗阻,因其弟弟有HAE病史,查补体C4为 0.093 g/L,后转至北京某医院查补体C1抑制因子为0.07 g/L,补体C4为 0.083 g/L,确诊为HAE。予达那唑0.2 g、2/d口服,2个月后复查补体C1抑制因子为0.11 g/L,补体C4 为0.126 g/L,因合并肝功能轻度异常,遂以达那唑0.2 g/d维持治疗。
1.3 家系调查 本文例1确诊为HAE后对其进行家系调查,发现家族共4代17人,其中6人罹患HAE,见图1。其母曾出现反复皮肤水肿,后因脑血管病去世。Ⅱ1 19岁时出现喉头水肿,因未及时抢救而死亡。Ⅱ2 30岁左右时出现口周水肿,每年发作1~4次,易在情绪激动或劳累后复发,症状持续1~2 d后自行缓解,因无其他临床症状,未就诊;5年前确诊为鼻咽癌、糖尿病,后未再出现口周水肿,亦未行补体C1抑制因子检测。Ⅱ4及Ⅲ4主要表现为急性腹痛。
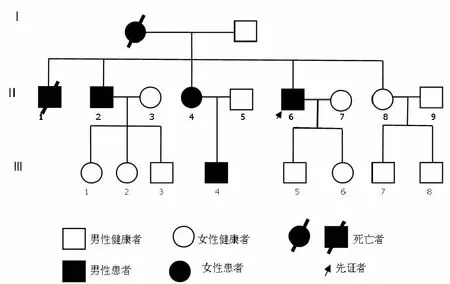
图1 遗传性血管性水肿家系调查图谱
注:Ⅰ~Ⅲ指1~3代;1~9为家系成员编号
2 讨论
2.1 发病机制 HAE由Osler[1]率先报道,发病率为1/10万~1/15万[2]。本文家系共3代15人,其中6人罹患HAE,且男女均发病,符合常染色体显性遗传特点,与相关文献[2-9]报道一致。其发病原因C1INH(一种α2球蛋白)减少或功能缺损,C1过度活化,C4及C2裂解失控,使生成的补体激肽增多,微血管通透性增高,从而引起水肿。HAE分为3种亚型[10],由于C1INH编码基因突变导致该蛋白产物功能缺陷,故HAE-I型及HAE-II型的C1INH含量和功能低下,且均小于诊断标准的50%,其中Ⅰ型(HAE-I型)又称普通型,占HAE的85%,血浆C1INH含量及功能均低下;Ⅱ型(HAE-II型)又称变异型,占HAE的15%,C1INH含量正常但功能低下。目前国内外报道的与HAE发病相关的C1INH基因突变多达上百种,有很强的异质性[11]。该家系有3例行C1INH基因检测,均符合HAE-I型的临床特点。Ⅲ型与C1INH基因突变无关,目前研究结果提示其与凝血因子FⅫ编码基因突变有关[12-13]。
2.2 临床特点 HAE主要表现为皮下或黏膜下水肿,可累及身体任何部位,以四肢、颜面、生殖器及消化道和呼吸道黏膜较为常见[14]。本文家系即出现皮肤、胃肠道和呼吸道损害。当呼吸道黏膜受累时,存在可致快速呼吸困难或窒息的潜在风险,是一种危及生命的急性水肿发作,病死率高达30%~40%[15-16]。该家系有2例出现呼吸道黏膜水肿,1例因就诊不及时死亡,1例曾行2次气管切开,确诊后予达那唑口服未再出现喉头水肿。该家系中Ⅱ4出现心前区疼痛及胸闷等类似冠脉粥样硬化性心脏病的表现,经冠脉造影确诊并经皮冠脉支架植入术后仍有胸痛反复发作,后出现脑血管症状,经综合治疗症状好转后又出现腹痛。目前文献[1-9,14-17]未见HAE心血管受累的报道,故患者经皮冠脉支架植入术后病情反复是否与HAE相关,有待进一步证实。
2.3 治疗 HAE的治疗分为急性发作期治疗和缓解期预防性治疗,后者包括短期预防性治疗和长期预防性治疗。目前发作期首选治疗方法为补充C1INH浓缩剂,推荐剂量为体重低于50 kg时静脉输注500 U,体重为50~100 kg时静脉输注1000 U,体重超过100 kg时静脉输注1500 U;同时还可选择新鲜冰冻血浆(FFP)静脉输注;若不能及时得到C1INH浓缩剂或FFP,可加大雄激素的剂量,同时予对症支持治疗。缓解期预防性治疗可选择作用较弱的雄激素维持。该家系3例确诊后均口服达那唑维持治疗,效果满意。
2.4 误诊原因分析及防范措施 HAE发病率低,临床表现复杂多样,医师对其认识不足,致长期诊断不明确,或仅做出症状诊断,加之多数医院不具备确诊条件,致误诊误治。提示临床遇及反复发作的局限性皮肤、口周和喉头水肿,或反复发作的不明原因腹痛,且腹痛可自行缓解的患者时,若体格检查无明显阳性体征,需警惕HAE的可能,及时行相关补体检查,争取早诊断、早治疗,以改善预后。
[1] Osler W. Landmark publication from The American Journal of the Medical Sciences: Hereditaryangioneuroticoedema. 1888[J].Am J Med Sci, 2010,339(2):175-178.
[2] Jimenez Saab N, Gomez Vera J, Lopez TiroJ,etal. Hereditary angioedema.Areport of a case and literature review[J].Rev AlergMex, 2006,53(1):34-41.
[3] 王印国,韦国强,陈娟,等.遗传性血管性水肿一例家系分析[J].临床误诊误治,2011,24(1):47-48.
[4] 严雪敏,张宏誉.间断呕吐、腹水-C1酯酶抑制物缺乏症一例报告[J].中华临床免疫和变态反应杂志,2009,3(1):63-65.
[5] 金春海,金京春,朴哲,等.以腹水为主要表现的遗传性血管性水肿1例[J].临床合理用药杂志,2009,2(6):12.
[6] 施和建,张国龙.遗传性血管性水肿一家系二例[J].中华医学遗传学杂志,2012,29(1):103.
[7] 袁强,周红雨.遗传性神经血管性水肿一家系五例[J].中华医学遗传学杂志,2007,24(1):120.
[8] 张淑环.遗传性血管性水肿1例及家系调查[J].临床皮肤科杂志,2004,33(2):113.
[9] 任华丽,张宏誉.133例遗传性血管性水肿患者的临床分析[J].中华医学杂志,2007,87(39):2772-2776.
[10]Gompels M M, Lock R J, Abinun M,etal. C1 inhibitor deficiency:consensus document[J].ClinExpImmunol,2005,139(3):379-394.
[11]徐迎阳,支玉香.C1抑制物基因突变提前形成终止密码子导致遗传性血管水肿[J].中华临床免疫和变态反应杂志,2013,7(2):125-128.
[12]Dewald G, Bork K. Missense mutations in the coagulation factor Ⅻ(Hageman factor)gene in hereditary angioedema with normal C1 inhibitor[J].BiochemBiophys Res Commun, 2006,343(4):1286-1289.
[13]Bork K, Wulff K, Meinke P,etal. A novel mutation in the coagulation factor 12 gene in subjects with hereditary angioedema and normal C1-inhibitor[J].ClinImmunol,2011,141(1):31-35.
[14]Agostoni A, Cicardi M. Hereditary and acquired C1-inhibitor deficiency: biological and clinical characteristics in 235 patients[J].Medicine(Baltimore),1992,71(4):206-215.
[15]Bygum A. Hereditary angio-oedema in Denmark:a nationwide survey[J].Br J Dermatol, 2009,161(5):1153-1158.
[16]Iwamoto K, Mihara S, Ikezawa Z,etal.National prevalence survey of hereditary angioedema in Japan [J].Arerugi, 2011,60(1):26-32.
[17]李健.遗传性血管性水肿1例[J].承德医学院学报,2015,32(1):75-76.
223800 江苏 宿迁,江苏省南京鼓楼医院集团宿迁市人民医院风湿免疫科
R596
B
1002-3429(2017)03-0023-03
10.3969/j.issn.1002-3429.2017.03.010
2016-12-05 修回时间:2017-01-05)
