锂离子电池正极材料LiCoPO4的合成和改性研究
2017-03-28陈桂敏盛锁江王双才胡博徐艳辉
陈桂敏,盛锁江,王双才,胡博,徐艳辉
(1.苏州大学物理与光电*能源学部 苏州 215006; 2.湖州创亚动力电池材料有限公司 湖州 31300)
1 前言
近年来,随着二次锂离子电池的不断发展,尤其是正极材料的发展,具有橄榄石结构的磷酸盐系列化合物LiMPO4(M=Fe、Mn、Co、Ni)已然在新能源材料领域成为研究的热点[1,2]。其中,LiFePO4优点诸多,成本低,容量高,循环性能优异,安全性好,吸引了众多研究者的眼球,现已经商业化[3]。但其缺点也比较明显,电势仅为3.4V(vs.Li+),所以能量密度低。另外,同系列的LiMnPO4电压平台为4.1V,但Mn3+存在姜一泰勒效应,使得LiMnPO4容量保持率较低;LiNiPO4的电压平台很高,在5.2V-5.4V之间,如此高的电压平台已超出常规电解液的承受范围。LiCoPO4为橄榄石结构,属正交晶系,Pnmb空间群,较高的理论容量,约为167mAh/g,相对于锂的电极电势为4.8V,能量密度高,理论能量密度为802Wh/kg,约为LiFePO4的1.35倍,约是LiCoO2的1.6倍[4],且在脱嵌锂过程中体积变化小[5],有望成为新一代高容量、高电压的锂离子电池正极材料[6]。但LiCoPO4较低的电子导电性低,约为10-15S/cm[7],较低的锂离子扩散率以及高电位使有机溶剂分解等缺陷,导致其倍率性能和循环性能较差[8],其应用受到很大的限制。
目前,除了选择合适的合成方法以及优化合成条件合成粒径较小且均匀的LiCoPO4材料[9],改善LiCoPO4正极材料电化学性能最有效的方法有:1、加入导电剂(如碳,Co2P,Fe2F);2、包覆碳,LiFePO4等;3、掺杂金属离子[10,11]。
2 LiCoPO4锂离子的拖脱嵌机理
Jörg J. Schneider等人[12]认为,LiCoPO4正极材料在充电时锂离子的脱出过程存在两步,第一个脱出平台在4.8V,第二个脱出平台在5.0V,而放电过程中表现出来的只有一步,还原平台在4.7V,其CV曲线如图1所示;Simon Theil[13]、Lucangelo Dimesso[14]以及M·K·Devaraju等人[15]的研究结果也证实了其两步机理。Hua Ju[16]等人通过高温固相法合成的LiCoPO4发现,纯的或碳含量很低的LiCoPO4材料在充电过程中也存在两个锂离子脱出过程,脱出平台分别为4.82V和4.92V,并提出此过程是一个三相机理,中间相为LixCoPO4(0.2≤x≤0.45),第一个平台是由LiCoPO4相转化为LixCoPO4,第二个平台是由LixCoPO4转化为CoPO4,而当样品中碳含量高时,材料的锂离子脱嵌机理为一步脱嵌机理,无中间相的产生。Wu Jun[17]等人也证明了杂质的存在对锂离子的脱嵌机理有相同的影响。Quang Duc Truong等人[18]用溶胶凝胶法合成的LiCoPO4材料也为两步脱嵌机理,第一个锂离子脱出平台在4.8V,但他认为第一个锂离子脱出平台是由LiCoPO4相转化为Li0.7CoPO4相过程,第二个锂离子脱出平台在4.9V,即由Li0.7CoPO4相转化为CoPO4相,同样,在放电曲线中只有一个平台。(如图1)目前,关于LiCoPO4正极材料两个充电平台准确解释仍存在争议。
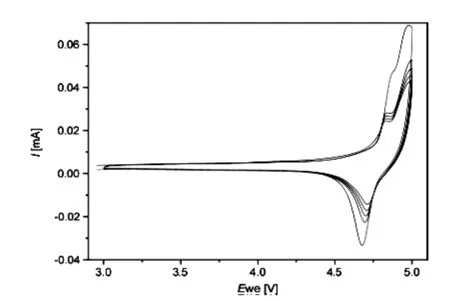
图1 温度700摄氏度下,60nm直径碳纳米管/磷酸钴锂复合材料的循环伏安图Fig.1 Cyclic voltammograms for a 3D CNT/LiCoPO4 composite comprising CNTs with a nominal diameter of 60 nm that has been processed at 700℃ in Air
虽然LiCoPO4正极材料的锂离子两步脱嵌机理得到了很多人的认可和证实,仍有一部分人却认为,LiCoPO4正极材料中锂离子的脱嵌机理为一步脱嵌机理。P.N.Poovizhi等人[19]采用溶胶凝胶法制备出纯相的LiCoPO4材料和碳包覆的LiCoPO4材料,CV曲线表明(如图2所示),包覆前后的样品均只有一对氧化还原峰,即只有一个充放电电压平台,提出了锂离子的一步脱嵌机理。Huanhuan Li等人[8]采用溶胶凝胶法合成的LiCoPO4和钇掺杂的LiCoPO4都只有一个氧化还原峰,证实了LiCoPO4中锂离子的一步脱嵌机理。
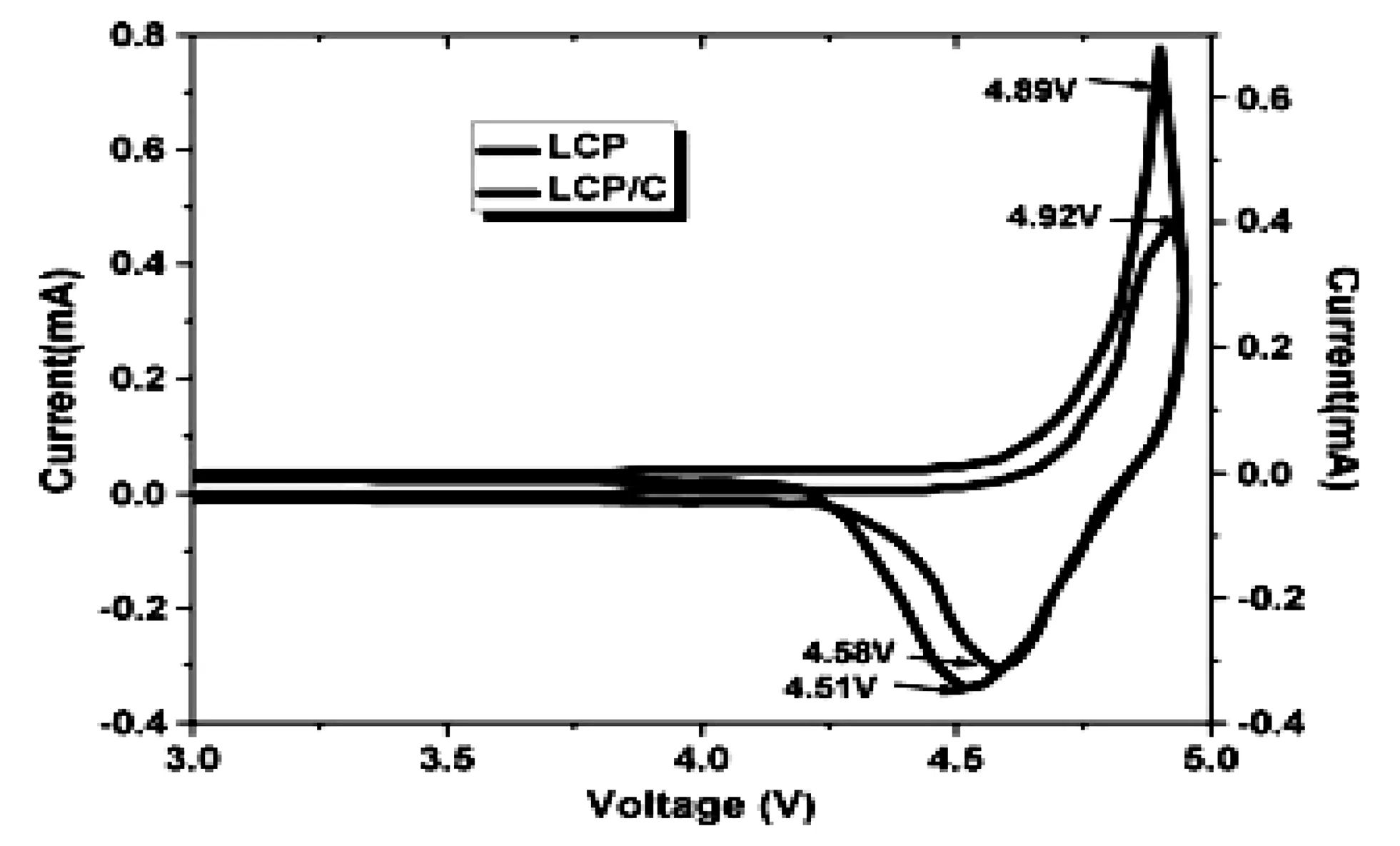
图2 温度700摄氏度下合成磷酸钴锂半电池和磷酸钴锂/石墨的循环伏安图 Fig.2 CV profile of LiCoPO4 heated at 700℃ and LiCoPO4/C
3 制备方法
目前,文献中合成LiCoPO4正极材料的方法比较多,如高温固相合成法,溶胶凝胶法,微波法,溶剂热法,其中,高温固相法在LiCoPO4合成占据主要位置,固相法合成的LiCoPO4材料可重复性和电化学性能都和优异,水热法和溶胶凝胶法合成的LiCoPO4材料容量在60mAh/g-70mAh/g,溶剂热法合成的LiCoPO4为纳米级粒子。本文就这几种方法介绍如下。
3.1 高温固相法
锂离子正极材料制备中最常用的方法就是高温固相法,该方法操作简便,易工业化生产,但该方法合成温度比较高,烧结时间长,能耗大,但效率较低,且合成材料均匀性较差。
J. Wolfenstine等人[20]以Li2CO3,CoC2O4·2H2O和NH4H2PO4为原料,经球磨后,分别在氧气和空气中经两段温度处理(375℃处理20h,775℃处理48h)后得到紫色的LiCoPO4,然后同样烧结条件在氩气中制得黑色的LiCoPO4。实验结果表明,在氩气中合成的LiCoPO4材料的电导率是在氧气和空气中合成的材料的电导率的104~105倍,且氩气中合成的LiCoPO4正极材料首次放电容量为100mAh/g,远大于在氧气和空气中合成的LiCoPO4材料。分析原因可能是由于在氩气中合成的LiCoPO4材料中存在第二相Co2P,提高了其电导率,从而电化学性能也得到改善。
Gangulibabu等人[21]也采用高温固相法以,Li2CO3,CoCO3和NH4H2PO4为原料,球磨后300℃下加热8h,然后在惰性气体氛围下700℃处理8h,LiCoPO4得到材料,再加入H2CO3+(NH4)2CO3后超声30min得到LiCoPO4/C纳米复合材料。XRD表明,材料为正交晶系,Pnmb空间群,且结晶良好。电化学测试表明5C下首次放电容量达123mAh/g,30圈后容量保持率为89%。分析原因可能是合成的样品中含有Co2P相以及导电碳,材料导电性大大提高,使循环性能和倍率性能良好。
Bo Jin等人[22]也采用高温固相法,以LiOH·H2O,CoO和(NH4)2HPO4为原料,研磨后热处理(750℃处理8h,350℃处理6h),经球磨后得到颗粒均匀的纳米级LiCoPO4正极材料,无杂质峰,且结晶性良好。
另外,I·C·Jang[23]和Wu Jun等人[17]也采用高温固相法合成了LiCoPO4正极材料。
3.2 溶胶凝胶法
溶胶凝胶法一般采用无机盐或者有机醇盐为原料,以水或者有机溶剂为溶剂得到溶胶,一定条件下得到凝胶,再经过干燥热处理得到产品。
Quang Duc Truong等人[18]采用溶胶凝胶法,以LiNO3,Co(NO3)2和NH4H2PO4为原料,以柠檬酸为螯合剂制得前驱体,80℃加热搅拌2h后升温至120℃,空气中处理2h后,500℃烧结5h得到去杂质相且结晶性良好的LiCoPO4正极材料。电化学测试表明,材料在0.1,0.2,0.5和1C下首次放电容量分别为117,93,80,和67mAh/g。透射电镜观察发现LiCoPO4中Co原子占据了Li原子位置,阻碍了锂离子的扩散,从而导致容量较低,倍率性能不好。
Lucangelo Dimesso[24]等人以Li(CH3COO)·2H2O,Co(CH3COO)2·4H2O,H3PO4和柠檬酸为原料,80℃下蒸发3h,得到均匀的凝胶,并分别在空气和氧气氛围中热处理。实验结果表明,在空气中热处理得到的LiCoPO4为单一晶相,大小不均匀的粒子粘结,CV曲线显示氧化峰和还原峰电势差较小表现出较好的可逆性。1/25C下首次放电容量仅为37mAh/g。而在氧气中得到的LiCoPO4中存在Li4P2O7和Co2P杂质相,粒子大小均匀,粒径约为2μm,但CV曲线显示其极化现象严重,可逆性较差,首次放电容量为42mAh/g,但容量保持率较好。
3.3 微波法
微波法具有升温速率快,加热均匀等优点,因此也常用作合成材料的一种便捷的方法。
Reginald E.Rogers等人[25]通过微波法,将化学计量比的LiOH,CO(CH3COO)2·4H2O溶于四甘醇中,搅拌30min,加如85%的H3PO4溶液和NH4OH,得到前驱体溶液,在微波合成器中2.45GHz,1200W下辐射5min,清洗,干燥得到 LiCoPO4,首次放电容量为128mAh/g,10圈后容量保持率为80%。
3.4 溶剂热法
M K Devaraju等人[15]以CoCl2·6H2O,H3PO4和LiOOCH3为原料,油酰胺为表面活性剂和还原剂,采用超临界溶剂热法,得到粒子宽度50nm-200nm,长度为100nm-300nm,厚度为5nm-15nm的C包覆的 LiCoPO4材料。XRD和XPS表明材料在400℃下结晶良好,产物纯净,无杂质相。充放电测试表明,首次放电容量为130mAh/g,10个循环后放电容量仍有89mAh/g,分析可能是由于较高的电压下电解液发生分解和脱锂相的不稳定导致容量衰减。
另外,R. Sharabi[26]和The Nam Long Doan等人[27]还采用水热法和喷雾热解法分别合成了LiCoPO4和LiCoPO4/C纳米复合材料。
4 LiCoPO4正极材料的改性研究
LiCoPO4材料锂离子扩散率和电子导电率较低,成为现在发展和应用的重要因素。对此,除了在采用不同的合成方法和优化制备工艺上改善LiCoPO4材料的性能外,加入导电剂,进行碳包覆以及金属离子掺杂等改性方法也能很好的改善LiCoPO4材料的电化学性能。
4.1 合成LiCoPO4/C复合材料
E. J. Kim等人[28]采用溶胶凝胶法,以LiNO3,Co(CH3COO)2·4H2O,NH4H2PO4,柠檬酸为原料,以葡萄糖为碳源合成了纯的LiCoPO4和 LiCoPO4/C复合材料。实验结果表明,400℃下烧结得到的LiCoPO4中存在杂质相Co3O4,而700℃下烧结得到的LiCoPO4为单相。700℃下烧结得到的LiCoPO4/C复合材料粒径在400nm-600nm。电化学测试显示,400℃下烧结得到的材料性能最佳,首次放电容量达144.6mAh/g,放电平台为4.7V,与纯 LiCoPO4材料相比,首次放电容量增加了44%,但容量衰减较快,当Li过量时,容量保持率得到提高。
Seung-Min Oh等人[29]采用沉淀法合成了 LiCoPO4/C复合材料,实验结果表明,当碳含量为5.wt%时,材料具有最好的电化学性能,在0.1C和5C倍率下,首次放电容量分别为145mAh/g和72mAh/g,且容量保持率较好。分析LiCoPO4性能得到改善的原因,采用NH4CoPO4·xH2O为前驱体,加热时有N2和H2生成,提供了一个很好的还原氛围,且乙炔黑均匀分布在LiCoPO4纳米粒子周围增加了其电子导电性,同时保护了材料免受电解液的氧化攻击,金属Co的存在也增加了电子导电性。
L.Dimesso等人[30]通过溶胶凝胶法合成了 LiCoPO4/纳米碳纤维复合材料,实验结果表明,730℃下处理5h得到的LiCoPO4材料为无杂质相,结晶良好,且均匀分布在纳米碳纤维表面,其含量为250.wt%-300.wt%。电化学测试表明,在0.1C倍率下,首次放电容量最大,为46mAh/g。
Lucangelo Dimesso等人[14]也采用溶胶凝胶法合成了LiCoPO4/泡沫碳复合材料,分别采用了氮气和空气量中不同的气体氛围。在氮气氛围中合成的LiCoPO4/泡沫碳复合材料中除LiCoPO4晶相,还存在Li4P2O7和Co2P等杂质相,在1/25C倍率下首次放电容量100mAh/g。现在空气中处理,然后在氮气中处理得到的LiCoPO4/泡沫碳复合材料除主要晶相LiCoPO4外,还存在少量的Co2P相,CV曲线表明其首次循环氧化还原峰分别在5.2V和4.6V,(如图3所示)1/25C倍率下首次放电容量为65mAh/g。因此在氮气中比在氧气中得到的LiCoPO4/泡沫碳复合材料容量高,分析原因是在氮气中得到的材料较高的碳含量且和杂质相的存在,增加了导电性,电化学性能也得到改善。
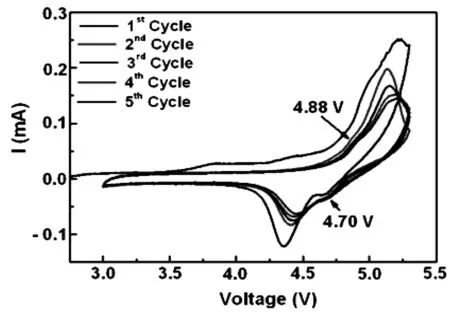
图3 氮气气氛下700摄氏度烧结12小时后得到的LiCoPO4/泡沫碳复合材料的循环伏安图(扫描速率0.1 mv/s,扫描电压2.5V-5.3V) Fig.3 Cyclic voltammograms for LCP/CNF after annealing in nitrogen at T=700℃ for t=12h(scan rate 0.1 mv/s,in the potential range 2.5V-5.3V vs. Li+/Li).
除了碳包覆外,Jang等人[23]采用LiFePO4对LiCoPO4进行包覆,实验结果表明,包覆后首次放电容量达132mAh/g,比LiCoPO4的高出19mAh/g,且循环性能明显提高,CV测试表明LiFePO4包覆后,材料可逆性良好。Jiangfeng Ni等人[22]还将Li4Ti5O12与LiCoPO4掺杂进行改性研究,也取得了良好效果。
4.2 金属离子掺杂
Huanhuan Li等人[6]采用溶胶凝胶法合成了 LiCoPO4和钇掺杂的LiCoPO4。XRD表明,纯的LiCoPO4和钇掺杂LiCoPO4的均为橄榄石结构,正交晶系,属Pnmb空间群,少量钇掺杂,峰尖锐且强度大,说明适量钇掺杂结晶更完美,当掺杂量大于 2.5.wt%时出现杂质相。SEM显示,钇掺杂后粒子半径减小且粒径均匀。电化学测试表明,当钇掺杂量为1%时,电化学性能最好,首次放电容量达153mAh/g,而纯LiCoPO4首次放电容量为130mAh/g,电压平台在4.8V,钇掺杂LiCoPO4比纯LiCoPO4的放电平台高且长。CV曲线显示,LiCoPO4和钇掺杂LiCoPO4均有一个氧化峰和还原峰,钇掺杂LiCoPO4比LiCoPO4的氧化还原峰电势差小,与充放电曲线一致。因此,通过钇掺杂,LiCoPO4正极材料的可逆性和锂离子扩散率得到明显提高,电化学性能得到改善。
Lucangelo Dimesso等人[32,33]用等价离子Ca2+,Mg2+取代LiCoPO4中的Co2+,得到LiCo0.9M0.1PO4(M=Ca,Mg)。实验结果表明,相同条件下合成的LiCoPO4,LiCo0.9Ca0.1PO4和 LiCo0.9Mg0.1PO4均为有序的橄榄石结构,且Ca2+和Mg2+均成功嵌入晶格中取代了Co2+,粒度较小。LiCoPO4中杂质相为CoP2O7,LiCo0.9Ca0.1PO4中杂质相为Co2P,Li3PO4,(Ca,Co)3(PO4)2, LiCo0.9Mg0.1PO4中有杂质相为Co2P。CV曲线表明,LiCoPO4, LiCo0.9Mg0.1PO4和LiCo0.9Ca0.1PO4放电平台分别为4.4V,4.6V和4.5V。(如图4所示)室温0.1C下首次放电容量分别为60mAh/g,68mAh/g和36mAh/g。阻抗测试显示,LiCoPO4经Ca和Mg取代后,阻抗减弱,锂离子扩散率增加。Lucangelo Dimesso还研究了不同量Mg取代后材料的电化学性能,结果表明,当Mg含量为2.5.wt%时首次放电容量最大,为88mAh/g,但容量衰减很快,且随Mg含量增加放电容量逐渐减少,但容量衰减减缓。
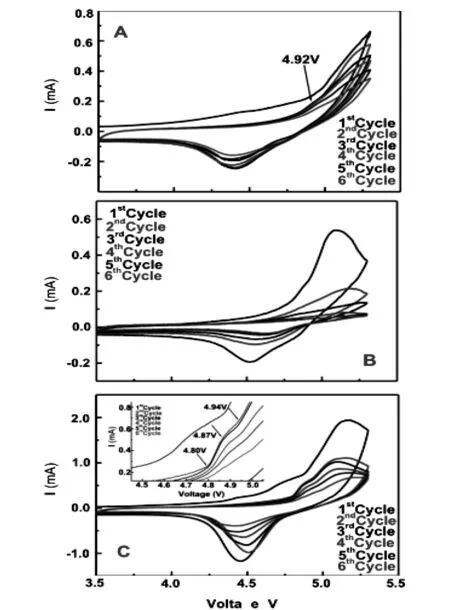
图4 (A)Co,(B)Mg,(C)Ca取代磷酸钴锂中Co前后的循环伏安图(扫描速率0.05mv/s,扫描电压范围2.5V-5.3V) Fig.4 Cyclic voltammograms recorded for the LiCo0.5M0.1P04 samples, (A)M=Co (B)M=Mg and (C)M=Ca respectively (scan rate 0.05 mv/s,in the potential range 2.5V-5.3V vs. Li+/Li).
Jan L. Allen等人[34]用Fe取代Co得到了 LiCo0.9Fe0.1PO4材料,实验结果表明,相同条件下合成的LiCoPO4和LiCo0.9Fe0.1PO4的粒子导电率分别为4×10-6S/cm和 1×10-5S/cm,电子导电率分别为8×10-16S/cm和2×10-12S/cm。因此,通过Fe取代,LiCoPO4材料的离子和电子导电性都得到了一定程度的提高。
另外,栗欢欢[35]和汪燕呜[36]等人分别用Mn和V取代来该改善LiCoPO4的性能,实验结果表明,通过这些金属离子取代后,LiCoPO4正极材料的导电性,放电容量以及循环性能都得到一定程度的改善。
5 结语
目前,如何提高锂离子电池的比容量,是锂离子电池的发展方向,这就对正极材料有较高的要求:首先,要有很好的脱嵌锂性;其次,更好的循环性能和高倍率特性。LiCoPO4正极材料的高电压、高理论容量,使其最有希望成为新一代高性能正极材料。然而, LiCoPO4正极材料的发展仍有很多阻力:1.材料的电子导电率较低;2.循环性能有待提高;高电压时电解液氧化分解。目前,除了以上文中提到的方法来提高LiCoPO4材料的性能外,开发出新一代耐高压、抗氧化的电解液[37,38]以及隔膜[26]也是一种有效应用LiCoPO4正极材料的好途径。如果人类能在这些方面得到突破,使 LiCoPO4电化学性能得到提高,那 LiCoPO4的应用前景将会更加广阔。
[1] Ehrenberga H, Bramnika N N, Senyshynb A, Fuessb H. Solid State Sciences, 2009, 11: 18-23.
[2] Xua J T, Shu L, Avdeevb M, Salea M, Liua H K, Dou S X. Electrochimica Acta, 2013, 88: 865-870.
[3] Ju H, Wu J, Xu Y H. J. Chem. Sci. Vol, 2013, 3(125): 687-693.
[4] Wolfenstine J, Read J, Allen J L. Journal of Power Sources, 2007, 163: 1070-1073.
[5] Jähne C, Neef C, Koo C, Meyerb H P, Klingelera R. Mater J. Chem. A, 2013, 1: 2856-2862.
[6] Julien C M, Mauger A. Ionics, 2013, 19: 951-988.
[7] Prabu M, Selvasekarapandian S, Reddy M V, Chowdari B V R. J Solid State Electrochem, 2012, 16: 1833-1839.
[8] Lia H H, Wang Y, Yang X L, Liu L, Chen L, Wei J P. Solid State Ionics, 2014, 255: 84-88.
[9] Tussupbayev R, Taniguchi I. Journal of Power Sources, 2013, 236: 276-284.
[10] Sarapulova A, Mikhailova D, Schmitt L A, Oswald S, Bramnik N, Ehrenberg H. J Sol-Gel Sci Technol, 2012, 62: 98-110.
[11] Dimesso L, Spanheimer C, Becker D, Jaegermann W. Journal of the European Ceramic Society, 2014, 34: 933-941.
[12] Schneider J J, Khanderi J, Popp A, Engstler J, Tempel H. A. Sarapulova act, Eur. J. Inorg. Chem, 2011: 4349-4359.
[13] Theil S, Fleischhammer M, Axmann P, Mehrens M W. Journal of Power Sources, 2013, 222: 72-78.
[14] Dimesso L,Cherkashinin G,Spanheimer C,Jaegermann W. Journal of Alloys and Compounds, 2012, 516: 119-125.
[15] Devaraju M K, Rangappa D, Honma I. Electrochimica Acta, 2012, 85: 548-553.
[16] Ju H, Wu J, Xu Y H. International Journal of Energy and Environmental Engineering, 2013, 4(22).
[17] Wu J, Li Z H, Ju L, Li D C, Zheng J W, Xu Y H. Rare Metal Materials and Engineering, 2013, 42(4): 0684-0687.
[18] Truong Q D, Devaraju M K, Tomai T, Honma I. Mater Interfaces, 2013, 5: 9926-9932.
[19] Poovizhi P N, Selladurai S. Ionics, 2011, 17: 13-19.
[20] Wolfenstine J, Lee U, Poese B, Allen J L. Journal of Power Sources, 2005, 144: 226-230.
[21] Gangulibabua,Nallathambya K,Meyrickb D,Minakshib M. Electrochimica Acta, 2013, 101: 18-26.
[22] Jin B, Gu H B, Kim K W. J Solid State Electrochem, 2008, 12: 105-111.
[23] Janga I C, Lima H H, Leea S B, Karthikeyana K, Aravindana V, Kangb K S, Yoonc W S, Chod W I, Leea Y S. Journal of Alloys and Compounds, 2010, 497: 321-324.
[24] Dimesso L, Jacke S, Spanheimer C, Jaegermann W. J Solid State Electrochem, 2012, 16: 911-919.
[25] Rogers R E, Clark G M, Matthew O N, Ganter M J, DiLeo R A, Staub J W, Forney M W, Landi B J. J Appl Electrochem, 2013, 43: 271-27.
[26] Sharabi R, Markevich E, Borgel V, Salitra G, Aurbach D, Semrau G, Schmidt M A, Schall N, Stinner C. Electrochemistry Communications, 2011, 13: 800-802.
[27] Doan N L, Taniguchi I. Journal of Power Sources, 2011, 196: 5679-5684.
[28] Kim E J, Xu H Y, Lim J S, Kang J W, Gim J H, Mathew V, Kim J. J Solid State Electrochem, 2012, 16: 149-155.
[29] Oh S M, Myung S T, Sun Y K. J. Mater. Chem, 2012, 22: 14932.
[30] Dimesso L, Spanheimer C, Jaegermann W, Zhang Y, Yarin A L. Electrochimica Acta, 2013, 95: 38-42.
[31] Ni J F, Liu W, Liu J Z, Gao L J, Chen J T. Electrochemistry Communications, 2013, 35: 1-4.
[32] Dimesso L, Spanheimer C, Jaegermann W. Journal of Power Sources, 2013, 243: 668-675.
[33] Dimesso L, Spanheimer C, Jaegermann W. Journal of Alloys and Compounds, 2014, 582: 69-74.
[34] Allen J L, Thompson T, Sakamoto J, Becker C R, Jowa T R, Wolfenstine J. Journal of Power Sources, 2014, 254: 204-208.
[35] 栗欢欢, 杨晓亮, 魏进平, 周震, 阎杰. 电化学, 2008, 2(14): 210-212.
[36] 汪燕呜, 王广健, 张茂林, 王阿呜. 功能材料, 2011, 4(42): 679-681.
[37] Sharabi R, Markevicha E, Fridman K, Gershinsky G, Salitra G, Aurbach D, Semrau G, Schmidt M A, Schall N, Bruenig C. Electrochemistry Communications, 2013, 28: 20-23.
[38] Markevich E,Sharabi R,Gottlieb H,Borgel V, Fridman K, Salitra G, Aurbach D, Semrau G, Schmidt M A, Schall N, Bruenig C. Electrochemistry Communications, 2012, 15: 22-25.
