高效液相色谱-荧光法测定牛羊组织中阿苯达唑及其代谢物的残留量
2017-03-16吴宁鹏彭丽孟蕾陈彦龙马浩轩河南省兽药饲料监察所郑州450008郑州大学郑州450000河南省郑州市第七高级中学郑州450008
吴宁鹏,彭丽,孟蕾,陈彦龙,马浩轩(.河南省兽药饲料监察所,郑州 450008; .郑州大学,郑州 450000; .河南省郑州市第七高级中学,郑州 450008)
高效液相色谱-荧光法测定牛羊组织中阿苯达唑及其代谢物的残留量
吴宁鹏1,彭丽1,孟蕾1,陈彦龙2,马浩轩3
(1.河南省兽药饲料监察所,郑州 450008; 2.郑州大学,郑州 450000; 3.河南省郑州市第七高级中学,郑州 450008)
建立了同时测定牛羊组织中阿苯达唑及其代谢物的高效液相色谱-荧光检测方法。试样中待测物经乙酸乙酯提取,酸性氧化铝固相萃取柱和MCX固相萃取柱净化,高效液相色谱-荧光法测定,以外标法定量。药物在5~2000 μg/L浓度范围内线性关系良好,相关系数均大于0.999。方法的检出限为0.025 mg/kg,定量限为0.05 mg/kg,在不同组织中添加浓度范围为0.05~10 mg/kg,其平均回收率为60.6%~119.1%,相对标准偏差在1.7%~19.1%之间。方法抗干扰能力强、灵敏度高,适用于牛羊组织中阿苯达唑及其代谢物残留量的测定。
牛羊组织;阿苯达唑及其代谢物;高效液相色谱-荧光法
阿苯达唑(ABZ)是一种苯并咪唑类驱虫药物,是抗虫性最强的驱虫药物之一。广泛用于动物的肠道寄生虫感染病,在我国兽医临床使用最广泛[1]。阿苯达唑在动物体内不稳定,动物口服摄入后,经肠道消化后会在较短时间内代谢为阿苯达唑亚砜(ABZSO)、阿苯达唑砜(ABZSO2)、阿苯达唑-2-氨基砜(ABZ-2NH2-SO2)[2]。动物毒理学表明阿苯达唑及其活性代谢物阿苯达唑亚砜具有致畸及胚胎毒性作用[3]。
由于一些养殖户为追求经济利益,违规使用而且长期、超量使用,从而使阿苯达唑及其代谢药物在动物组织中残留,因此,世界食品法典委员会和世界各国对动物源性食品中阿苯达唑药物残留均有严格的限量要求,国际贸易中也对其限量有严格规定[4]。我国规定在牛、猪、羊、鸡等动物的肌肉、脂肪、肝、肾等食品中阿苯达唑及其代谢药物最高残留限为0.05~1.0 mg/kg[5]。国内外对阿苯达唑及其代谢物检测分析方法报道较多,主要包括免疫检测法(ELISA)[6]、高效液相色谱法(HPLC)[2,7-10]、液相色谱串联质谱法(HPLC-MS/MS)[11-12]和气相质谱联用法(GC-MS/MS)[13]等。其中,HPLC-FLD法因具有检测成本低、灵敏度高等优点,被广泛应用于阿苯达唑及其代谢物的检测。目前牛羊组织中阿苯达唑及其代谢药物残留的检测方法中,存在空白干扰严重、灵敏度低、适应性差等问题,因此,本文对牛羊组织中阿苯达唑及其代谢物药物残留的样品前处理法进行了改进,减小基质干扰,从而提高检测的准确度,从源头上保障食品安全。
1 材料与方法
1.1 仪器与试剂 高效液相色谱仪配荧光检测器(Waters公司);XP205分析天平(瑞士Mettler Toledo 公司);N-EVAP-112氮吹仪(美国Organomation);SIGMA 3-30K离心机(德国SIGMA公司); MS3 basic涡旋混合器(IKA)。阿苯达唑、阿苯达唑砜、阿苯达唑亚砜,含量均大于98.3%,购于Dr. Ehrenstorfer公司,阿苯达唑氨基砜对照品,购于德国WITEGA;Waters Oasis MCX 固相萃取柱(6mL/150 mg);Supelco酸性氧化铝固相萃取柱(3 mL/1 g)。甲醇、乙腈、乙酸铵(色谱纯);二甲基亚砜(DMSO)、碳酸钠、焦亚硫酸钠、乙酸乙酯、正己烷、盐酸、乙二胺四乙酸二钠、氨水、冰醋酸(分析纯);实验用水为经Milli-Q净化系统制备的去离子水。
0.5 mol/L乙酸铵溶液:取1.927 g乙酸铵,用水溶解并稀释至500 mL,用冰醋酸调节pH至5.0。
1.2 标准溶液的配制 准确称取阿苯达唑、阿苯达唑砜、阿苯达唑亚砜和阿苯达唑氨基砜标准品各10 mg,分别于100 mL容量瓶中,用乙腈:二甲基亚砜(9:1)溶解并定容,配制成浓度为0.1 mg/mL的阿苯达唑、阿苯达唑砜、阿苯达唑亚砜和阿苯达唑氨基砜标准贮备液,-20 ℃以下保存,有效期1个月。
1.3 样品前处理 称取试样(2±0.02)g于50 mL离心管中,加2%的碳酸钠溶液0.5 mL, DMSO 1 mL和0.004 g/mL焦亚硫酸钠溶液溶液1 mL,涡旋1 min,加入乙酸乙酯10 mL,涡旋1 min,振荡5 min, 10000 r/min离心5 min,取上清液。残渣再加入乙酸乙酯10 mL,涡旋1 min,振荡5 min, 10000 r/min离心5 min,取上清液。合并两次上清液,40 ℃氮吹至近干,用正己烷5 mL复溶,加入乙腈∶0.2 mol/L盐酸(V∶V/9∶1)2 mL溶液,涡旋1 min,静置分层,弃去上层正己烷,余液作为备用液1。
酸性Al2O3固相萃取柱依次用乙腈∶0.2 mol/L盐酸(V∶V/9∶1)溶液3 mL、1 mol/L乙二胺四乙酸二钠溶液3 mL活化,取备用液1过柱,收集流出液,用乙腈∶0.2 mol/L盐酸(V∶V/9∶1)1 mL溶液淋洗,合并流出液,加0.2 mol/L盐酸溶液8 mL,混匀作为备用液2。MCX柱依次用甲醇3 mL、1 mol/L乙二胺四乙酸二钠溶液3 mL、水3 mL、0.2 mol/L盐酸溶液3 mL活化,取备用液2过柱,待全部液体流出后,依次用0.2 mol/L盐酸溶液3 mL、甲醇3 mL淋洗,抽干1 min,10%氨化乙腈溶液5 mL洗脱,收集洗脱液,于40 ℃水浴氮气吹干。用乙腈∶甲醇∶0.5 mol/L乙酸铵溶液(V∶V∶V/2∶2∶8)1 mL溶解残余物,过0.22 μm滤膜后供高效液相色谱测定(上机溶液应在72 h内完成测定)。
1.4 液相色谱参考条件 色谱柱:Waters Xbridge C18 (4.6×150 mm, 5 μm);流速1.0 mL/min;柱温30 ℃;进样量20 μL;流动相A为乙腈,B为甲醇,C为0.5 mol/L乙酸铵溶液,梯度洗脱程序见表1。激发波长290 nm,发射波长330 nm。
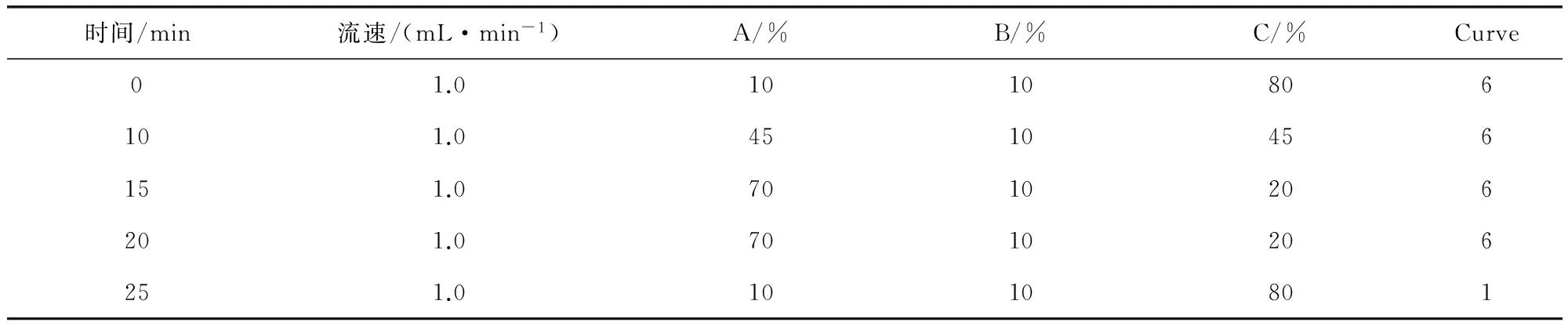
表1 流动相梯度洗脱条件
2 结果与分析
2.1 线性范围 准确移取阿苯达唑、阿苯达唑砜、阿苯达唑亚砜和阿苯达唑氨基砜标准溶液液适量,用乙腈∶甲醇∶0.5 mol/L乙酸铵(V∶V∶V/2∶2∶8)溶液稀释,配置成一系列浓度的混合标准溶液,从低浓度到高浓度依次进样,以色谱峰平均峰面积为纵坐标,对应浓度为横坐标,绘制标准曲线,各药物的回归方程及相关系数见表2。

表2 药物回归方程及相关系数
2.2 方法的灵敏度 添加适量混合标准溶液于2 g空白样品中,经提取后测定,依据信噪比S/N>3(按PtP算),确定阿苯达唑及其代谢药物的检出限均为0.025 mg/kg。依据信噪比S/N>10(按PtP算),确定阿苯达唑及其代谢药物的定量限均为0.05 mg/kg。2.3 方法的准确度和精密度考察 本方法考察了在牛肉、牛肝、羊肉、羊肝中的添加回收情况。采用标准加入法,在空白样品中添加0.05、2.5、5、10 mg/kg 4个不同浓度的阿苯达唑及其代谢物标准品,进行准确度和精密度试验,各浓度进行5个样品平行试验,重复3次,求相对标准偏差,计算结果见表3。
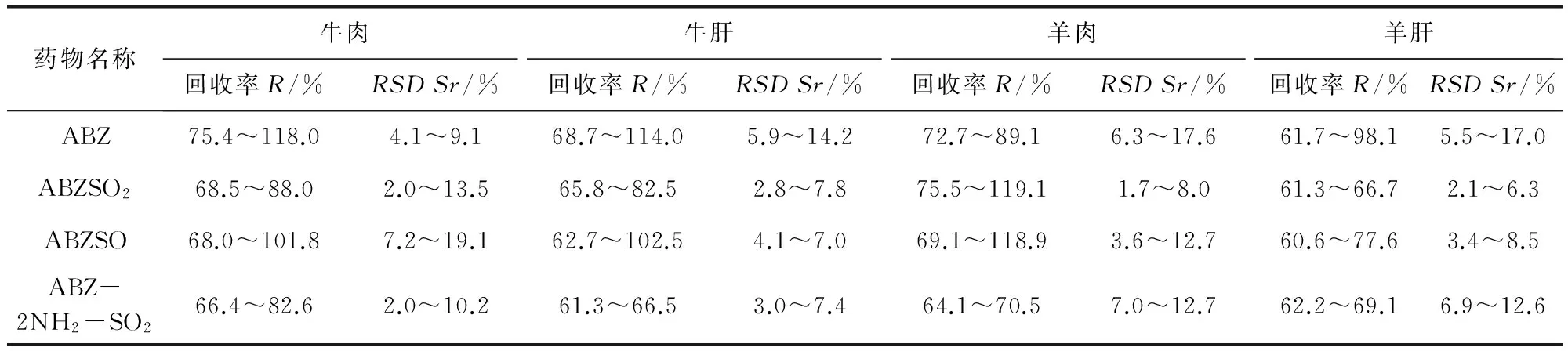
表3 牛、羊组织中阿苯达唑及代谢物回收率和相对标准偏差
从表中可以看出,本方法在空白牛羊组织中进行0.05~10 mg/kg的添加,平均回收率在60.6%~119.1%之间,批间相对标准偏差在1.7%~19.1%之间。说明该方法有较好的准确度与精密度。阿苯达唑及其代谢物标准溶液、空白及添加色谱图分别见图1~图3。
3 讨论与小结
3.1 仪器条件 阿苯达唑及其代谢物属于苯并咪唑类物质,既有紫外吸收又有荧光吸收。因此,按照参考文献方法分别选用紫外检测器和荧光检测器作为阿苯达唑及其代谢物的检测,实验结果表明选用荧光检测器对动物组织样品测定时,杂质干扰小,准确度高。因此,选择荧光检测器作为动物组织中阿苯达唑及其代谢物的检测器。
阿苯达唑类药物残留检测常用的流动相体系有甲酸-乙腈和乙酸铵-乙腈-甲醇等流动相体系,试验过程中对两种流动性体系进行了比较,最终确定了用0.05 mol/L乙酸铵-乙腈-甲醇(80∶10∶10)作为流动相进行梯度洗脱,流速1.0 mL/min,能够很好的控制阿苯达唑氨基砜、阿苯达唑亚砜、阿苯达唑砜和阿苯达唑的保留时间,避免了出峰较早易受到杂质峰干扰,也克服了保留时间较长而导致色谱峰展宽。
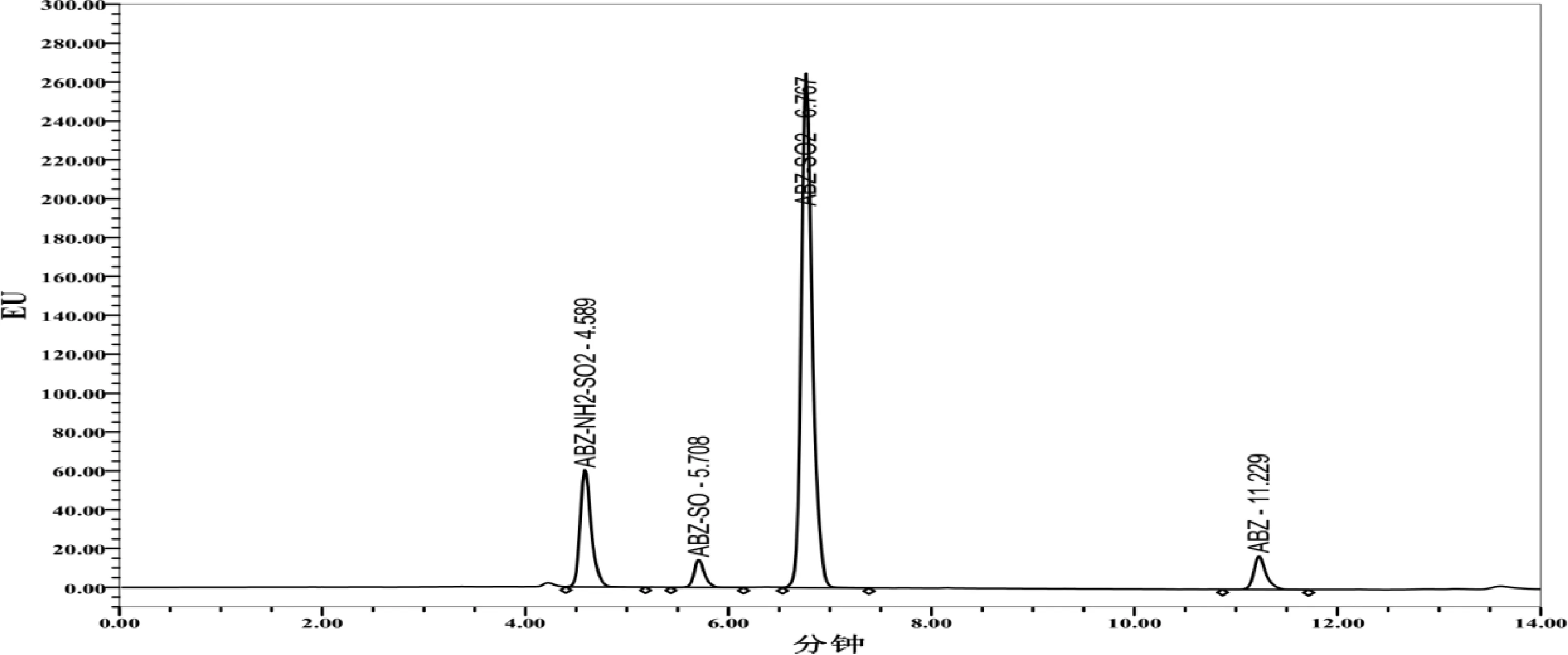
图1 阿苯达唑及其代谢物标准溶液色谱图(100 μg/L)
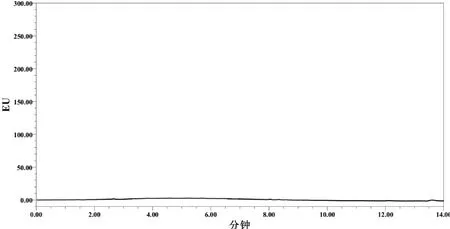
图2 羊肝组织空白试样色谱图
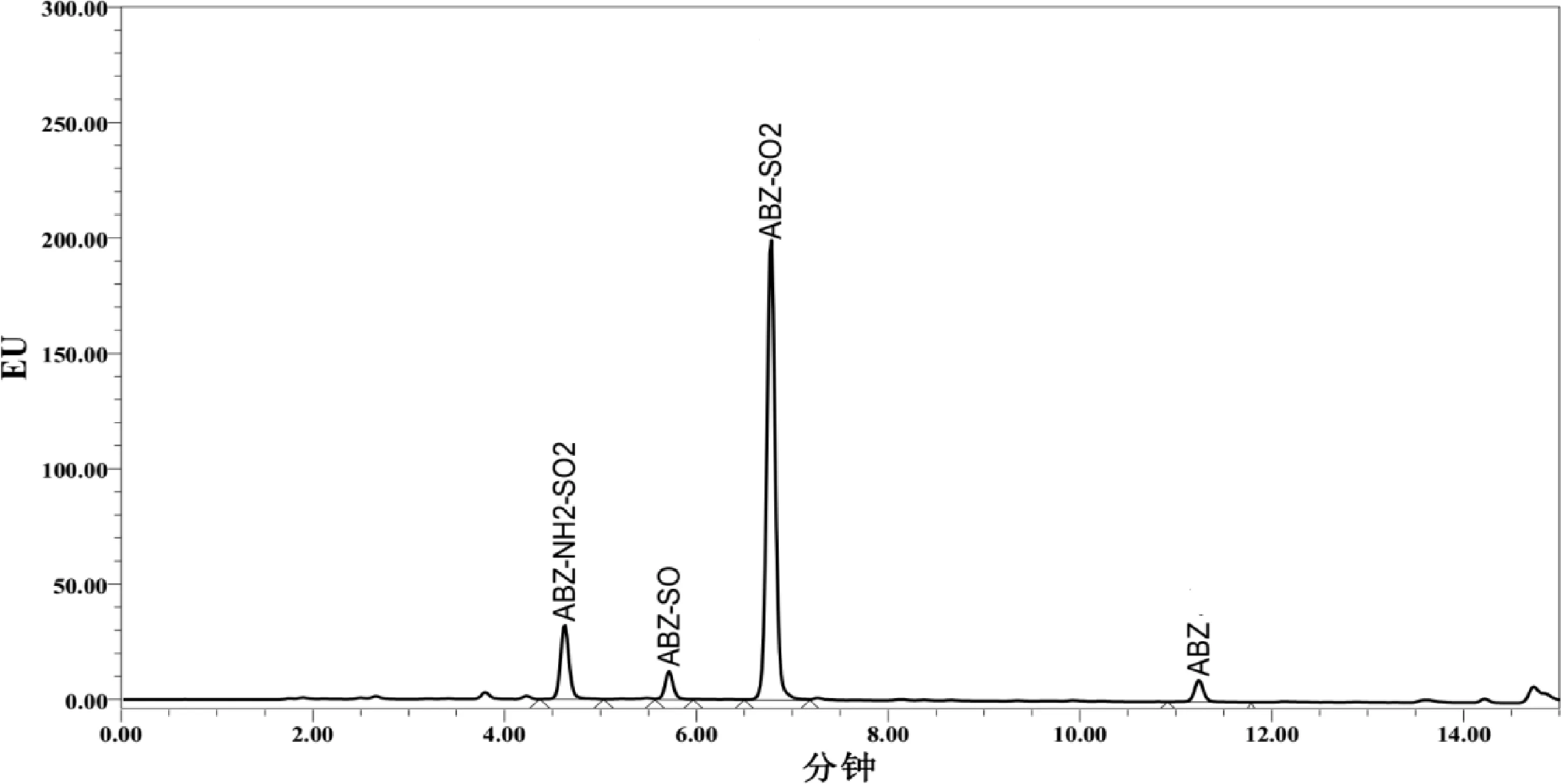
图3 羊肝组织空白添加阿苯达唑及其代谢物色谱图(0.05 mg/kg)
3.2 提取条件 本方法参考了相关文献和检测方法,提取液分别用乙腈、乙酸乙酯和乙腈+乙酸乙酯(V∶V/3∶1)为提溶剂进行5次平行提取实验。由实验结果可知,对于四种药物的提取效率为乙酸乙酯>乙腈+乙酸乙酯>乙腈,同时用乙酸乙酯作为提取溶剂时杂质干扰明显低于乙腈。因此,选用乙酸乙酯为动物组织中阿苯达唑及其代谢物的提取溶剂。由于阿苯达唑及其代谢物具有弱碱性,在不同的盐溶液和pH 条件下电离度不同,所以参考了国内外一些文献在乙酸乙酯提取溶液中加入5% NaOH、10% Na2SO4和2%Na2CO3,实验结果发现乙酸乙酯提取溶剂中加入0.5 mL 2%Na2CO3使提取液呈弱碱性,在该条件下阿苯达唑及其三种代谢物均有较高的回收率。为了减小基质对阿苯达唑及其代谢药物检测的干扰,在提取时加入1 mL抗氧化剂Na2S2O5(4 mg/mL)。
3.3 净化方法 参照文献的净化方法,先后比较了Sep-Pak C18(6 mL 150 mg)、Oasis MCX(6 mL 150 mg)、Oasis HLB(6 mL 150 mg)、ProElut SCX(6 mL 150 mg)固相萃取柱,结果表明MCX回收率最高。但由于样品基质比较复杂,仅过MCX柱净化,杂质干扰严重,尤其组织样品中极性化合物和阴离子化合物会影响动物组织中痕量的阿苯达唑及其代谢物的检出。因此,最终选用酸性Al2O3和MCX固相萃取柱作为净化柱。
该方法具有较好的灵敏度、准确度和精密度,适用于牛羊组织中阿苯达唑及其代谢物残留量的测定。
[1] 周丽萍. 动物组织中阿苯达陛代谢物多残留检测方法的研究[D]. 济南:山东大学,2009.
[2] 张小军, 郑斌, 张虹, 等. 超高效液相色谱-串联质谱法测定草鱼肉中阿苯达唑及其代谢物残留[J]. 分析化学,2011,39:815-820.
[3] Botsoglou N A,Fletouris D J. Drug residues in foods,pharmacology,food safety and analysis[M].New York:Marcei,Dekker Inc.,2001:269-298.
[4] Couneil Regulation No. 2377/90 and Commission Regulation Nos. 508/1999, 2385/1999, 2393/1999 and 807/2001[S].
[5] 李俊锁, 邱月明, 王超.兽药残留分析[M]. 上海:上海科学技术社, 2002: 459-495.
[6] 伏旭,李培武, 贺莉,等.酶联免疫吸附检测法的应用研究进展[J]. 氨基酸和生物资源, 2012, 34: 41-44.
[7] Dowling G, Cantwell H, O’Keeffe M,etal. Multi-residue method for the determination of benzimidazoles in bovine liver[J]. Anal Chim Acta, 2005, 529: 285-292.
[8] Moreno L, Lopez-Urbina M T, Farias C,etal. A high oxfendazole dose to control porcine cysticercosis: Pharmacokinetics and tissue residue profiles[J]. Food and Chemical Toxicology, 2012, 50: 3189-3825.
[9] Lange H, Eggers, Bircher J. Increased Systemic Availability of Albendazole when Taken with a Fatty Meal[J]. Eur J Clin Pharmacol, 1988,34:315-317.
[10]张素霞, 沈建忠,丁双阳,等. 牛肝中苯并咪唑类药物残留的高效液相色谱检测方法[J]. 中国兽药杂志, 2005, 39: 18-21.
[11]郭强,常孝勇. 动物组织中苯并咪唑类药物检测的LC-MS/MS法研究[J]. 河南农业科学,2012,41:152-156.
[12]Balizs G. Determination of benzimidazole residues using liquid Chromatography and tandem mass speetrometry[J]. J Chromatogr B,1999,727:167-177.
[13]Markus J, Sherma J. Gas-chromarographic-mass spectrometric confirmatory Method for albendazole residues in cattle liver[J].J AOAC Int,1992,75:1135-1137.
(编辑:侯向辉)
Determination of Albendazole and Its Main Metabolites Residues in Cattle and Ovine Tissues by HPLC-FLD
WU Ning-peng1,PENG Li1,MENG Lei1,CHEN Yan-long2,MA Hao-xuan3(1.HenanInstituteofVeterinaryDrugandFeedsControl,Zhengzhou450008,China;2.ZhengzhouUniversity,Zhengzhou450000,China;3.HenanProvinceZhengzhouNo.7MiddleSchool,Zhengzhou450008,China)
A method based on high performance liquid chromatography with fluorescence detector (HPLC-FLD) has been developed for the determination of albendazole and its main metabolites residues in cattle and ovine tissues. The analytes were extracted with ethyl acetate, and cleaned by Alumina-A and MCX solid phase extraction cartridges. Quantification of the analytes were achieved by HPLC-FLD using external standard method. The calibration curves of the drugs were linear in the concentration ranges of 5~2000 μg/L with correlation coefficients more than 0.999. Detection limits and quantification limits of the method for the analytes were 0.025 mg/kg and 0.05 mg/kg in cattle and ovine tissues. The average recoveries ranged from 60.6% to 119.1% at the spiked level of 0.05~10 mg/kg in tissues. The relative standard deviation was in the range of 1.7%~19.1%. With the advantages of good anti-interference ability and sensitivity, the method adapts to the determination of albendazole and its main metabolites residues in cattle and ovine tissues.
cattle and ovine tissues; albendazole and its main metabolites;HPLC-FLD
2016-10-10
A
1002-1280 (2017) 02-0035-05
S859.84
2016年国家畜禽产品未知危害因子识别与已知危害因子安全性评估项目(GJFP2016007)
吴宁鹏,高级兽医师,从事兽药质量监测与监督研究。E-mail:wnppeking2002@163.com
